Letzten Monat wurde ich eingeladen, am Center for Mendelian Genomics (CMG) Analysis and Methods Development Meeting über „Population-based estimation of penetrance in rare disease“zu sprechen. Hier ist die Blog-Post-Version meines Vortrags.
was ist Penetranz und warum kümmern wir uns?
Penetranz ist die Wahrscheinlichkeit, bei einem bestimmten Genotyp eine bestimmte Krankheit zu entwickeln., Man kann von altersabhängiger Penetranz sprechen, also der Prozentsatz der Menschen mit dem Genotyp, die die Krankheit im Alter entwickeln 40, im Alter 50, und so weiter; Ich spreche normalerweise in Bezug auf das Lebensrisiko, was die Wahrscheinlichkeit bedeutet, dass Sie jemals die Krankheit entwickeln, bevor Sie sterben. Inhärent darin ist, dass das lebenslange Risiko bei Krankheiten, die bei Erwachsenen auftreten, niemals 100% betragen kann, da Sie immer zuerst an etwas anderem sterben könnten.
Penetranz ist für Personen, die sich prädiktiven Gentests unterziehen, äußerst wichtig — die erste Frage vieler Menschen lautet: „Bedeutet das, dass ich definitiv an der Krankheit erkranken werde?”., Es ist jedoch oft sehr schwierig, eine feste Schätzung der Penetranz zu erhalten.
traditionelle Methoden zur Schätzung der Penetranz
In einer idealen Welt besteht der richtige Weg zur Schätzung der Penetranz darin, von Geburt an eine große Kohorte von Menschen mit einem bestimmten Genotyp festzustellen, ihnen zu folgen, bis alle an etwas oder anderem gestorben sind, und dann zu fragen, wie viele jemals die Krankheit entwickelt haben, bevor sie gestorben sind. Da die Genotypisierungstechnologie vor weniger als einem menschlichen Leben erfunden wurde, wurde dies nie für eine Krankheit getan.
Stattdessen verwenden Forscher häufig familienbasierte Methoden, um die Penetranz abzuschätzen., Eine typische Studie würde jeden betrachten, der mit dem gegebenen Genotyp beobachtet wurde, und fragen, wie viele diease haben, oder wie viele Krankheit durch ein bestimmtes Alter haben. Familienbasierte Methoden leiden unter allgegenwärtigen Ermittlungsvorurteilen ., ermittelt auf der Grundlage der Präsentation mit Krankheit
Als Beispiel für diesen letzten Punkt wählen bei genetischen Prionenkrankheiten nur 23% der Risikopersonen prädiktive Gentests, und in Stammbaumdaten, die ich hatte
Alle oben aufgeführten Faktoren arbeiten in die gleiche Richtung und neigen dazu, die Penetranzschätzung aufzublasen.
Forscher sind sich dieser Probleme seit langem bewusst und haben einige Lösungen vorgeschlagen. Als ein Beispiel umfasst die Kin-Kohortenmethode die zufällige Ermittlung gesunder Individuen aus einer Population, die Genotypisierung, die Anamnese und den Vergleich der Überlebenskurven ihrer Verwandten ersten Grades., Dies ist eine sehr clevere Lösung, die jedoch darauf beruht, eine ausreichend große Anzahl von Menschen mit einem krankheitsverursachenden Genotyp ermitteln zu können, ohne das Vorhandensein einer Krankheit festzustellen. Es funktionierte also für BRCA1-und BRCA2-Varianten bei amerikanischen aschkenasischen Juden , aber für viele seltenere genetische Bedingungen ist es unpraktisch, weil Sie Zehntausende oder Hunderttausende von Menschen rekrutieren müssten, um sogar eine Person mit einem Genotyp von Interesse zu finden.,
populationsbasierte Methoden
Aus allen oben beschriebenen Gründen ist es sehr nützlich, orthogonale, populationsbasierte Methoden zu haben, um Fragen zur Penetranz zu stellen. Die erste wichtige Erkenntnis dabei ist, dass eine vollständig penetrante genetische Variante in der Bevölkerung nicht häufiger auftreten sollte als die Krankheit, die sie verursacht. Die Anwendung dieser Logik in der Praxis bedeutet, dass Sie auch für ungewöhnliche Varianten gute Schätzungen der Allelfrequenz benötigen, und das war bis vor kurzem schwer zu bekommen. ExAC, eine Datenbank genetischer Variationen in 60.706 menschlichen Exomen, bietet neue Möglichkeiten ., Viele Personen in ExAC wurden als Fälle oder Kontrollen für verschiedene häufige, komplexe Krankheiten festgestellt, aber keine wurde für Mendelsche Krankheit festgestellt, so ExAC ist eine gute Referenzdatenbank für die Untersuchung der meisten genetischen Krankheiten.
Durch die Bereitstellung von Allelfrequenzinformationen in der Allgemeinbevölkerung hat ExAC, wie frühere Referenzdatenbanken wie ESP, deutlich gemacht, dass die klinische Genetik ein großes Problem hat: Viele Varianten, von denen berichtet wird, dass sie genetische Krankheiten verursachen, verursachen keine genetische Krankheit oder zumindest nicht die meiste Zeit.,
Zwei Datenbanken-HGMD und ClinVar-sammeln Aussagen aus der Literatur und aus klinischen Labors, die besagen, dass eine bestimmte genetische Variante eine bestimmte genetische Erkrankung verursacht. Bei der letzten Zählung gab es über 100.000 einzigartige Berichte über krankheitsverursachende genetische Varianten in diesen Datenbanken. Die Durchschnittliche person in ExAC hat 54 von Ihnen . Offensichtlich hat die durchschnittliche Person nicht wirklich 54 genetische Krankheiten., Natürlich wird ein Großteil dieses Überschusses durch eine kleine Anzahl wild hochfrequenter Varianten verursacht, die offensichtlich keine genetische Erkrankung verursachen, und vieles davon kann Berichten zufolge rezessive Varianten sein, die in einem heterozygoten Zustand in ExAC gefunden werden. Aber selbst wenn wir nur Varianten in dominanten Krankheitsgenen mit einer Allelfrequenz von <1% betrachten , sehen wir immer noch 0,89 angeblich pathogene Varianten pro Person, und es ist eindeutig nicht der Fall, dass ~90% der Menschen eine dominante genetische Krankheit haben., Im gesamten Allelfrequenzspektrum gibt es also viele angeblich pathogene Varianten, die nicht so pathogen sind. Als Anne O ‚ Donnell und ich uns die angeblich pathogenen Varianten mit den höchsten Allelfrequenzen in ExAC ansahen und fragten, wie sie es geschafft hatten, als pathogen eingestuft zu werden, stellten wir fest, dass das Problem meistens auf ein Papier in der Literatur zurückging, das aufgrund unzureichender Beweise einen Anspruch auf Pathogenität erhoben hatte.
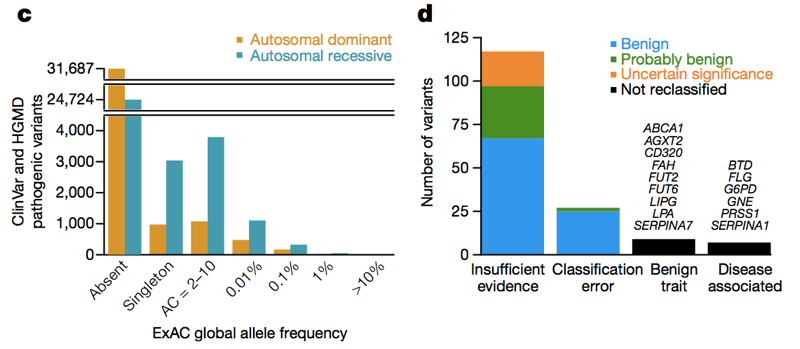
Oben: Figuren 3C und 3D aus ., Über das Allelfrequenzspektrum und sowohl in dominanten als auch in rezessiven Krankheitsgenen gibt es viele Berichten zufolge pathogene Varianten, die in ExAC auftreten. Von hoher (>1%) Frequenz berichten zufolge pathogene Varianten, einige sind wirklich pathogen, einige sind wirklich merkmalsbezogen, aber das Merkmal ist gutartig, und einige sind Fehler der Annotation in Datenbanken-aber die Mehrheit basiert auf Literatur mit unzureichenden Beweisen.,
Informationen zur Allelfrequenz von ExAC haben es nun ermöglicht, über 200 genetische Varianten von pathogen zu gutartig, wahrscheinlich gutartig oder von ungewisser Bedeutung umzuklassifizieren . Diese Art von Reklassifikationen lösen manchmal einen Rückstoß von den ursprünglichen Autoren aus, die vorgeschlagen haben, dass eine Variante eine genetische Krankheit verursacht, die argumentieren könnte, dass eine Variante immer noch pathogen sein könnte, aber mit unvollständiger Penetranz. Aber wie „unvollständig“ kann unvollständige Penetranz sein?, Wir müssen quantitativ werden, denn wenn das Lebenszeitrisiko höchstens 1% beträgt, ist es dann immer noch vernünftig zu sagen, dass eine Variante eine genetische Krankheit „verursacht“ oder „pathogen“ist? Während Allelfrequenzinformationen niemals beweisen können, dass eine Variante keine Assoziation mit einer Krankheit hat, kann sie Grenzen setzen, was die mögliche Penetranz sein könnte, und in vielen Fällen ist es sogar für ziemlich seltene Varianten möglich zu zeigen, dass es keine Möglichkeit gibt Eine Variante verleiht ein Risiko irgendwo in der Nähe von 100%.,
Um quantitativ zu werden, müssen wir unsere frühere Beobachtung erweitern — dass eine vollständig penetrante genetische Variante in der Bevölkerung nicht häufiger sein sollte als die Krankheit, die sie verursacht. Dies ist alles einfache Mathematik und Populationsgenetik, aber es wird zu oft nicht in der Praxis angewendet. Hier sind zwei Möglichkeiten, wie wir über die Allelfrequenz nachdenken können, wenn wir Rückschlüsse auf Pathogenität und Penetranz ziehen.
maximale glaubwürdige Allelfrequenz
Angenommen, Sie untersuchen das Exom eines Patienten mit Mendel-Krankheit und versuchen, die ursächliche Variante zu identifizieren., Mein Kollege James Ware hat eine Strategie entwickelt, um dieses Exom gegen die Allelfrequenzinformationen in ExAC zu filtern und dabei die folgende Logik zu nutzen., Die maximale Allelfrequenz, die für eine Variante plausibel ist, um eine dominante genetische Erkrankung zu verursachen, entspricht der Prävalenz der Krankheit mal der allelischen Heterogenität (Anteil der Fälle , die einer Variante zuzuordnen sind) geteilt durch Penetranz (weniger penetrante Varianten können häufiger sein), geteilt durch 2 (weil wir diploid sind):
\
Zum Beispiel verursacht Prionenkrankheit bei 1 von 5.000 Todesfällen, und die häufigste Variante (E200K) wird in 5% der Fälle gefunden, so dass eine 100% penetrante Variante möglicherweise keine Allelfrequenz haben kann, die größer ist als als 0,0005% (1 in 200,000) ., Kardiomyopathie betrifft 1 von 500 Menschen, die häufigste Variante findet sich in <2% der Fälle, so dass eine 50% ige Penetrantenvariante keine Allelfrequenz größer als 0,004% haben kann . Die Formel für rezessive Krankheiten ist eine Kerbe komplizierter, aber James hat es auch ausgearbeitet und es wird beschrieben in .
Während Menschen in der Vergangenheit häufig Varianten mit einer Allelfrequenz herausgefiltert haben >0.1% wenn wir versuchen , die Ursache einer dominanten Krankheit zu identifizieren, können wir tatsächlich viel strenger sein., Der Nachteil ist, dass bei niedrigen Allelzahlen unsere Fähigkeit, die Allelfrequenz abzuschätzen, durch Abtastvarianz begrenzt ist. Wenn wir uns zum Beispiel Varianten ansehen, die bei einer Allelfrequenz von 1% bei Europäern in ESP zu sehen sind, haben diese Varianten auch bei ExAC-Europäern eine Frequenz von etwa 1%. Varianten mit einer Frequenz von 0,1% in ESP sind jedoch in ExAC etwas seltener, und die meisten Singletons (Varianten, die genau einmal in ESP zu sehen sind) tauchen in ExAC nicht ein zweites Mal auf.
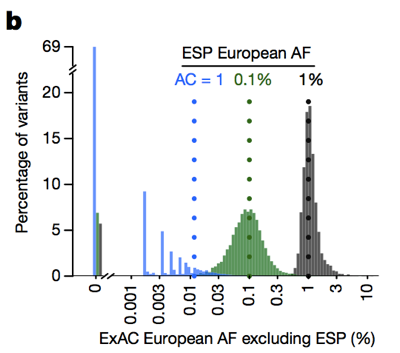
Oben: Abbildung 3B aus . Je niedriger die Allelanzahl ist, desto weniger gut ist eine Schätzung der Allelfrequenz.,
Je niedriger die Allelzahl, desto konservativer müssen wir daher sein. Wir haben ein Framework entwickelt, um dies mit der 95% igen Obergrenze der Poisson-Verteilung zu tun, wie viele Allele bei einer bestimmten Häufigkeit beobachtet werden können, und haben vorberechnete Werte für alle ExAC (verfügbar auf FTP), die Sie verwenden können-lesen Sie mehr über die Methoden in . James hat auch diese praktische Web-App erstellt, mit der Sie herausfinden können, was die „maximale glaubwürdige Allelfrequenz“ für Ihre Krankheit von Interesse sein sollte.,
Inhärent in diesem Ansatz ist, dass je niedriger die Penetranz einer Variante ist, desto höher ist die Häufigkeit, die sie in der allgemeinen Bevölkerung haben könnte. Aber Sie müssen auch herausfinden, dass, wenn die Penetranz ziemlich niedrig ist, sagen wir, weniger als 10%, dann ist der klinische Nutzen dieser Variante auch gering. James und Nicky Whiffin haben Daten vorgelegt, die zeigen, dass fast der gesamte klinische Nutzen der Sequenzierung bei Kardiomyopathie von Varianten mit einer Häufigkeit von <0.001% stammt — häufigere Varianten tragen kumulativ wenig, wenn überhaupt, Risiko bei .,
Schätzung und Grenzen des Lebenszeitrisikos
Denken Sie daran, dass Penetranz die Wahrscheinlichkeit einer Erkrankung bei einem bestimmten Genotyp ist. Oder, wenn wir ein Allel eher als genotypisches Modell betrachten, die Wahrscheinlichkeit einer Krankheit ein bestimmtes Allel gegeben. Wir können dies als P(D|A) schreiben. Sobald wir dies tun, wird klar, dass nach Bayes‘ Theorem
\
Jeder dieser Begriffe eine bestimmte Bedeutung hat:
Beachten Sie hier, dass „Populationskontrollen“ eine Gruppe bedeuten, die weder für das Vorhandensein noch für das Fehlen der Krankheit ausgewählt wurde. Nur ein Teil der Bevölkerung.
Also:
\
Diese Logik ist nichts neues., Die Verwendung von Bayes ‚ Theorem zur Schätzung des Krankheitsrisikos geht zumindest auf die Schätzung des Krebsrisikos bei Rauchern zurück , und seine Anwendung auf die Genetik wurde fast so lange in Betracht gezogen . Aber damit diese Gleichung für seltene Krankheiten funktioniert, benötigen Sie ziemlich gute Schätzungen der Fall-und Populationskontrollallelfrequenz, und diese waren bis vor kurzem schwer zu bekommen. Dank ExAC gibt es immer mehr Situationen, in denen diese Gleichung relevant ist.
Hier ist der R-Code, den ich (ursprünglich hier) geschrieben habe, um die Penetranz basierend auf dieser Formel zu schätzen.,
Wenn Sie den R-Code nicht selbst ausführen möchten, hat James Ware ihn auf der Registerkarte „Penetrance“ dieser Web-App implementiert, sodass Sie Ihre Nummern einfach in Ihren Browser einfügen können.
Um 95% Konfidenzintervalle auf Penetranz zu schätzen, habe ich den Ansatz von übernommen . Sie geben die Allelanzahl (AC) und die Anzahl der Individuen (N) für Fälle und Kontrollen ein, und die Obergrenze der 95% CI wird basierend auf der oberen 95% CI der Binomialverteilung für die Fallallelfrequenz und der unteren 95% CI für Kontrollen berechnet., Umgekehrt basiert die untere Penetranzgrenze auf der unteren Grenze der Fallallelfrequenz und der oberen Grenze der Regelallelfrequenz. Sie könnten zu Recht behaupten, dass die resultierenden Konfidenzintervalle größer sind, als sie sein sollten, da diese Formel 95% CIs für beide Allelfrequenzwerte verwendet. Sie könnten auch zu Recht behaupten, dass die Binomialverteilung bei niedrigen Allelzahlen aufgrund der in Abbildung 3B dargestellten Verzerrung kein guter Schätzer ist (und ich würde diese Formel sicherlich niemals auf Singletons — Varianten anwenden, die nur einmal in ExAC beobachtet wurden)., Aber am Ende des Tages, aus Gründen, die ich näher am Ende dieses Beitrags diskutieren werde, wird diese Formel wirklich am besten verwendet, um einen Ballpark zu erhalten, Größenordnung Schätzung der Penetranz. Wenn Sie nach einer äußerst genauen Punktschätzung der Penetranz suchen, wird dieser gesamte Ansatz wahrscheinlich sowieso nicht für Sie funktionieren.
Wenn Sie die Gleichung neu anordnen, ist eine andere Möglichkeit, darüber nachzudenken:
\
Dies bedeutet, dass das erhöhte Risiko bei Menschen mit einem Genotyp proportional zum Verhältnis von Fall zu Population ist Kontrollallelfrequenz., Eine Variante, die das Risiko um das 200-fache erhöht, sollte in Fällen 200-mal häufiger auftreten als in der Allgemeinbevölkerung. (Beachten Sie, dass sich dieses Verhältnis von Allelfrequenzen geringfügig von Odds ratio unterscheidet, obwohl die beiden Kennzahlen für sehr seltene Varianten konvergieren.)
Anwendung prion-Krankheit
Wir gingen durch diese Logik in einer Studie, die wir veröffentlicht früher in diesem Jahr, Quantifizierung Penetranz von prion-Krankheit-Varianten ., Ich interessiere mich aus einem persönlichen Grund für die Prionenkrankheit — meine Frau birgt eine pathogene Variante in PRNP—, aber es stellt sich heraus, dass die Prionenkrankheit auch ein großartiger Testfall ist, um die obige Logik zur Schätzung der Penetranz zu verwenden. Keine der Personen in ExAC v1 wurde bei neurodegenerativen Erkrankungen festgestellt, daher ist ExAC wirklich ein guter Populationskontrolldatensatz für Prionenkrankheiten. Und weil Prionenkrankheiten „meldepflichtig“ sind, haben nationale Überwachungszentren eine außergewöhnlich gute Fallermittlung, und dank ihrer Großzügigkeit beim Datenaustausch konnten wir einen Datensatz von 10,460 sequenzierten Fällen sammeln.,
Wir fanden heraus, dass die>60 Varianten, von denen berichtet wurde, dass sie eine Prionenkrankheit verursachen, kumulativ 52 Allele in ExAC aufweisen. Das bedeutet, dass fast 1 von 1.000 Menschen eine dieser Varianten hat, und daher sind diese Varianten kumulativ viel häufiger als alle Prionenkrankheiten (die ~1 von 5.000 Todesfällen verursachen), geschweige denn alle genetischen Prionenkrankheiten (nur ~15% der Fälle sind genetisch bedingt). Dies ist genug, um uns zu sagen, dass nicht alle dieser Varianten möglicherweise vollständig penetrant sein können. Um festzustellen, welche Varianten die Schuldigen waren, haben wir die Fallserien verglichen., Varianten mit ausgezeichnetem vorherigen Nachweis der Pathogenität (Mendelsche Segregation und Mausmodelle) waren in Fällen üblich und fehlten in ExAC, was mit vollständiger oder nahezu vollständiger Penetranz übereinstimmte. Der größte Teil der überschüssigen Allelzahl in ExAC wurde durch Varianten beigetragen, die in Fällen ungewöhnlich waren und schwache vorherige Anzeichen von Pathogenität aufwiesen — diese Varianten sind wahrscheinlich gutartig oder tragen nur ein geringes Risiko bei. Mindestens drei Varianten erschienen intermediär, da sie bei Kontrollen für eine vollständige Penetranz zu häufig waren und dennoch in Fällen über Kontrollen angereichert waren.,
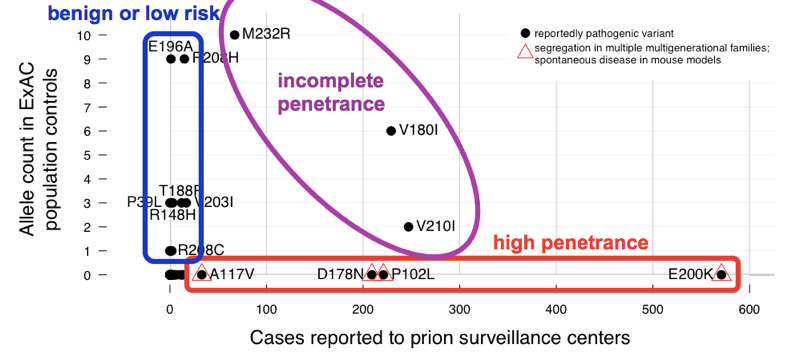
Oben: eine kommentierte version von Abbildung 2 aus .
Als wir die Penetranz für jede Variante unter Verwendung der obigen P(D|A) – Formel schätzten, stellten wir fest, dass es ein ganzes Penetranzspektrum für PRNP-Varianten gibt.
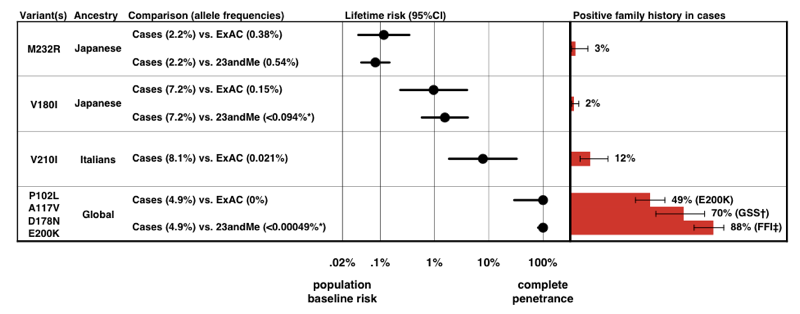
Oben: Abbildung 3 aus .
Beachten Sie die Skala auf der x-Achse — für eine Krankheit, die so selten ist, dass die vorherige Wahrscheinlichkeit, sie zu entwickeln, nur 0,02% beträgt, selbst ein 50-facher Risikoanstieg beträgt nur 1% Lebenszeitrisiko., Beruhigend ist, dass die Penetranzschätzungen, die wir allein aus Allelfrequenzinformationen ableiten, ziemlich gut mit dem Anteil der Fälle übereinstimmen, die eine positive Familienanamnese aufweisen.
Diese Arbeit hat bereits zu einer Änderung der Prognose für einige Personen geführt, denen ursprünglich geraten worden war, dass sie ein Risiko für Varianten mit hoher Penetranz haben-siehe auch Haydens Artikel über ExAC. Sie können meine und Sonia persönliche Reise mit dieser Studie hier lesen.,
Anwendung auf NR1H3
Multiple Sklerose (MS) ist eine komplexe Erkrankung mit vielen genetischen Risikofaktoren , es ist jedoch keine mendelsche Form der Krankheit bekannt. Anfang dieses Jahres berichtete eine Studie, dass eine Missense — Variante in einem nuklearen Hormonrezeptor — NR1H3 R415Q-die allererste mendelsche Form von MS verursacht . Diese Behauptung basierte auf einer dominanten Segregation mit Krankheiten in zwei Familien, aber der LOD — Score lag nur 2.2-unter der Schwelle für die genomweite Signifikanz in Familienverknüpfungsstudien, die eher 3.0 oder 3.6 entspricht . Und die fragliche Variante hat eine Allelfrequenz von 0.,031% der nicht-finnischen ExAC-Europäer. Das klingt vielleicht nicht nach einer hohen Allelfrequenz, aber es erweist sich als viel zu hoch für diese Variante, um Mendelsche MS zu verursachen .
Bedenken Sie, dass MS ein Lebenszeitrisiko (in der Allgemeinbevölkerung) von 0, 25% bei Frauen und 0, 14% bei Männern aufweist . Wenn 0,06% der Menschen in der Allgemeinbevölkerung R415Q-Heterozygoten sind und wenn sogar die Hälfte von ihnen MS entwickelt, würde diese Variante allein 0,03% der Bevölkerung ausmachen, die MS entwickelt.Wenn also insgesamt 0,25% der Menschen MS entwickeln, dann sollten etwa 12% von ihnen diese Variante haben., Stattdessen wurde die Variante nur bei 1 von einer Fallserie von 2,053 MS-Patienten gefunden .
Dies funktioniert auf eine Allelfrequenz von 0.024% in Fällen oder 0.049%, wenn wir zulassen, dass 2 Fälle in der Fallreihe gezählt werden. Dies ist nicht signifikant höher als die Frequenz in ExAC. Aber wenn diese Variante MS verursacht, sollte es in Fällen häufiger sein — viel häufiger. Erinnern Sie sich an unsere umgestaltet früheren Formel: P(D|A)/P(D) = P(A|D)/P(A). Dies bedeutet, dass eine Variante, die das Risiko um das X-fache erhöht, bei Kontrollen X-mal häufiger auftreten sollte. Also, wenn das Grundrisiko von MS 0 ist.,25% – und diese Variante ist 50% penetrant, es sollte 50/.25 = 200-fach häufiger in Fällen als Kontrollen. Wenn es sogar 10% Penetranz hatte, sollte es immer noch 10/sein.25 = 40 mal häufiger in Fällen als bei Kontrollen. Alternativ können Sie in Bezug auf Odds Ratios statt Wahrscheinlichkeiten denken. Das 0,25% ige Lebenszeitrisiko in der Allgemeinbevölkerung bedeutet 1: 399 Chancen, und wenn R415Q 50% Lebenszeitrisiko verleiht, wäre das 50:50 Chancen. (50/50)/(1/399) = 399, das Odds Ratio für R415Q müsste also 399 sein, damit diese Variante 50% Penetranz hat.,
Wenn wir stattdessen unsere Formel unter Verwendung des R-Codes von früher anwenden, ein Ausgangsrisiko von 0, 25% annehmen und die Berechnung auf 2 Allelen auf 2,053 Fällen im Vergleich zu 21 Allelen bei 33,369 ExAC-Personen stützen, stellen wir fest, dass die Obergrenze des 95% – CI bei der Penetranz 2, 2% beträgt. Selbst wenn R415Q mit dem MS-Risiko assoziiert wäre, könnte es nicht mehr als 2,2% Lebenszeitrisiko für die Entwicklung von MS bedeuten .,
In ihrer formalen Antwort und in PubMed haben die Autoren einen Vergleich mit LRRK2 G2019S bei der Parkinson-Krankheit angestellt, bei dem alle einig sind, dass es pathogen ist, das aber auch in ExAC vorkommt und nur eine bescheidene Odds Ratio hat, geschätzt auf 9.6 . Für diese Variante klappt die Mathematik. Die Parkinson-Krankheit ist mindestens um eine Größenordnung häufiger als MS, wobei das Lebenszeitrisiko zwischen 3, 7% und 6, 7% geschätzt wird . Diese Größenordnung größere Prävalenz bedeutet, dass die ~10-fache Anreicherung, die beobachtet wurde — LRRK2 G2019S in etwa gefunden 0.,1% der Kontrollen und 1% der Fälle-entspricht in etwa dem gemeldeten Lebenszeitrisiko von ~32% für Parkinson, das durch diese Variante übertragen wird . Diese quantitativen Details sind wichtig und für jede Variante und jede Krankheit unterschiedlich. Aus diesem Grund sind die in diesem Beitrag diskutierten Formeln nützlich, obwohl sie nur sehr grobe Schätzungen liefern und mehreren Einschränkungen unterliegen, wie unten erläutert.
Vorbehalte
In beiden oben beschriebenen Anwendungen wurden Allelfrequenzinformationen verwendet, um eine grobe Schätzung der Penetranz zu erhalten., In der Prionen-Krankheit, konnten wir zeigen, dass Varianten bisher vermutet, höchst penetrant übertragen lebenslange Risiko mehr in der Größenordnung von 0.1%, 1%, oder 10%. In der NR1H3-Geschichte reichten Allelfrequenzinformationen aus, um zu zeigen, dass die angeblich kausale Variante nicht mehr als ein paar Prozent Lebenszeitrisiko bergen konnte.
Der Versuch, Allelfrequenzdaten zu verwenden, um eine genauere Schätzung der Penetranz zu erhalten, wäre jedoch sehr schwierig. Zum Beispiel haben familienbasierte Studien über die Penetranz von PRNP E200K nicht einverstanden, mit Schätzungen im Bereich von 60% bis 90% Lebenszeitrisiko ., Seit die Prion-Studie herauskam, haben mich einige Leute aus E200K-Familien gefragt, ob ExAC-Daten helfen können, einzugrenzen, wo das Risiko in diesem Bereich liegt. Die Antwort ist leider nicht.
Hier sind die wichtigsten Gründe, warum ich denke, dass alle Penetranzschätzungen basierend auf der Allelfrequenz mit einem Körnchen Salz genommen werden müssen:
- Wenn eine Variante stark penetrant ist, ist es schwierig, eine Fallreihe zu erhalten, die keine verwandten Personen enthält. Wenn Ihre Fallserie Verwandte hat, haben Sie technisch keine unvoreingenommene Schätzung von P(A|D).,
- Wenn eine Krankheit tödlich ist, ist es schwierig, eine Populationskontrollreihe zu erhalten, die bei Menschen mit Varianten, die diese Krankheit verursachen, nicht zumindest etwas erschöpft ist. Dann haben Sie auch keine unvoreingenommene Schätzung von P (A).
- Vergleiche der Fall – und Kontrollallelfrequenz sind anfällig für Verwechslungen durch Bevölkerungsschichtung. In der Prion-Studie hatten wir keine genomweiten SNP-Daten zu Fällen, daher gab es keine Möglichkeit, dies perfekt zu kontrollieren.,
- Viele kausale Varianten für seltene Krankheiten sind so selten, dass wir selbst bei ExAC noch keine hinreichend genauen Schätzungen der Allelfrequenz haben, um eine bessere als eine grobe Antwort zu geben.
Bei allem, was gesagt wird, ist die populationsbasierte Allelfrequenzschätzung immer noch ein guter Weg, um grobe Größenordnungen der Penetranz zu erhalten und Vernunftprüfungen durchzuführen, ob eine genetische Variante plausibel für eine seltene Krankheit ursächlich sein könnte.