Rna-sequenciação (RNA-seq) tem uma ampla gama de aplicações, e não há um gasoduto ideal para todos os casos. Revisamos todos os principais passos na análise de dados RNA-seq, incluindo controle de qualidade, alinhamento de leitura, quantificação de níveis de gene e transcrição, expressão diferencial de gene, análise funcional e análise avançada. Eles serão discutidos mais tarde.
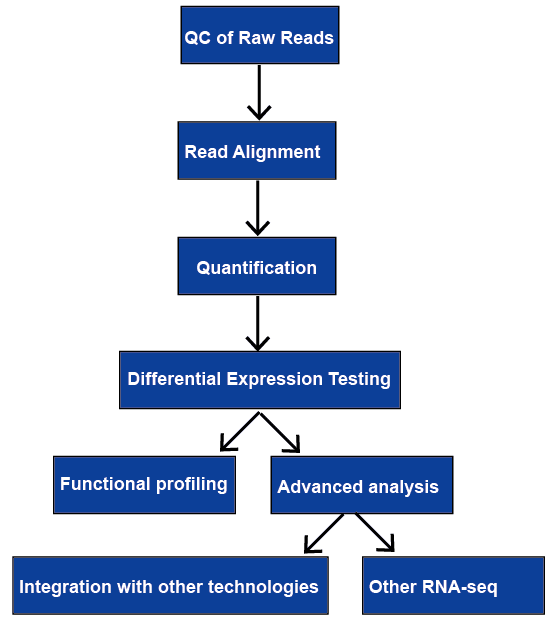
Figura 1. The general workflow of RNA-seq analysis.,
controlo de qualidade das leituras em bruto
controlo de qualidade das leituras em bruto RNA-seq consiste na análise da qualidade da sequência, do conteúdo de GC, do conteúdo do adaptador, dos k-Mer sobre-representados e das leituras duplicadas, dedicadas à detecção de erros de sequenciação, contaminações e artefactos de PCR. A qualidade de leitura diminui para o fim de leitura de 3′, bases com baixa qualidade, portanto, eles devem ser removidos para melhorar a mappability., Além da qualidade dos dados brutos, o controle de qualidade das leituras brutas também inclui a análise do alinhamento de leitura (uniformidade de leitura e conteúdo de GC), quantificação (viés, biótipos e contagens baixas) e reprodutibilidade (correlação, análise de componentes principais e efeitos de lotes).Tabela 1. The tools for quality control of RNA-seq raw reads.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Independentemente de uma referência de genoma ou transcriptoma estar disponível, as leituras podem mapear de forma única ou ser atribuídas a múltiplas posições na referência, que são referidas como leituras ou multi-mapeadas. Multireads genômicos são geralmente devido a sequências repetitivas ou domínios compartilhados de genes paralógicos. O multi-mapeamento transcriptoma surge mais frequentemente devido a isoformas genéticas. Assim, a identificação e quantificação da transcrição são desafios importantes para os genes alternativamente expressos., Quando uma referência não está disponível, RNA-seq lê são montados de novo usando pacotes como SOAPdenovo-Trans, Oases, Trans-ABySS, ou Trinity. As leituras específicas e longas de PE strand são preferidas, uma vez que são mais informativas. Tecnologias emergentes de leitura longa, como a sequenciação de PacBio SMRT e a sequenciação de Nanopore, podem gerar transcrições de comprimento completo para a maioria dos genes.
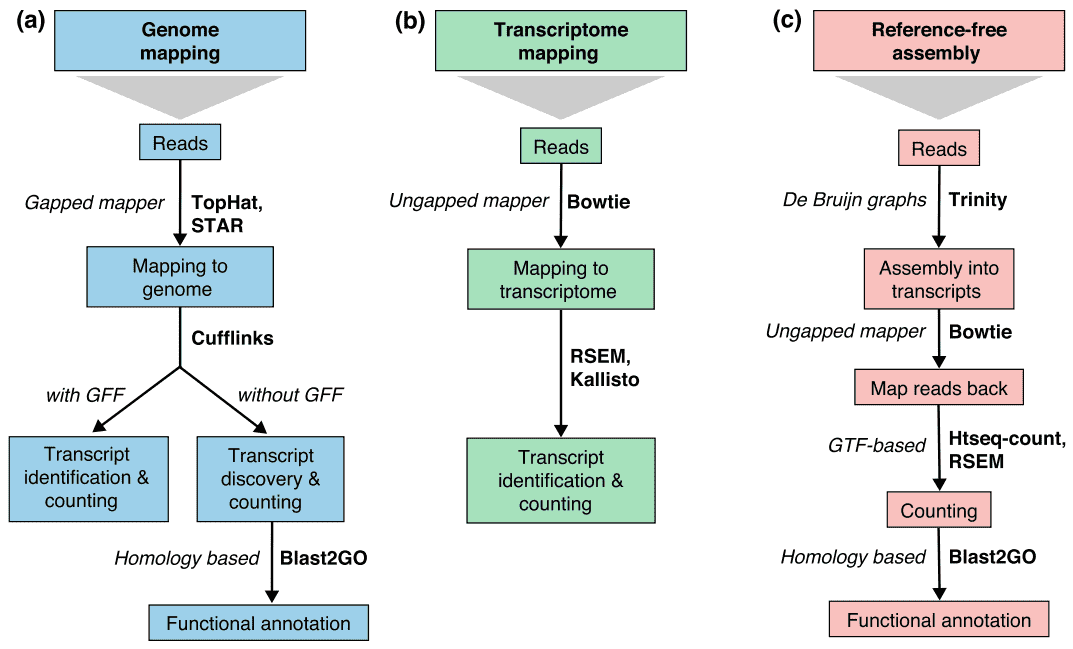
Figura 2. Três estratégias básicas para o mapeamento RNA-seq read (Conesa et al. 2016)., Abreviaturas: GFF, General Feature Format; GTF, gene transfer format; RSEM, RNA-seq by Expectation Maximization.
Tabela 2. The comparison of genome-based and de novo assembly strategies for RNA-seq analysis.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| botão de Punho | Projetado para tirar vantagem do PE lê, e pode usar GTF informações para identificar expressa transcrições, ou pode-se inferir transcrições de novo a partir do mapeamento dos dados. |
| RSEM | Quantificar a expressão do transcriptoma de mapeamento.Allocate multi-mapping reads among transcript and output within-sample normalized values corrected for sequencing biases., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
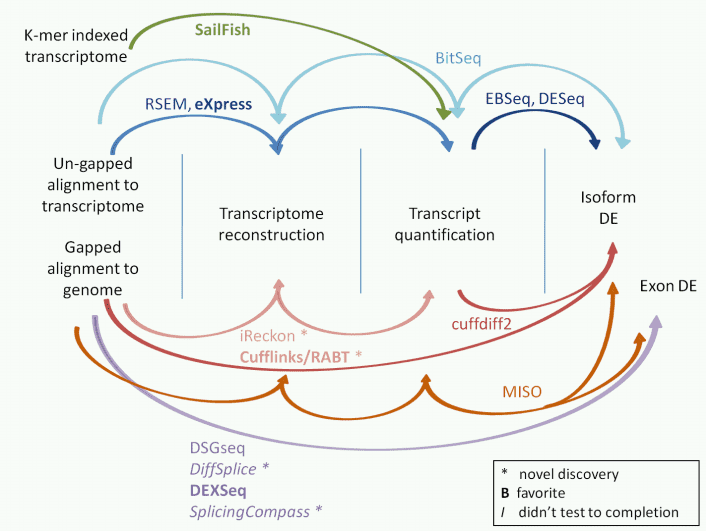
Figure 3. The tools for isoform expression quantification.,
Teste de expressão diferencial
Teste de expressão diferencial é usado para avaliar se um gene é expresso diferencialmente em uma condição em comparação com o outro(s). Os métodos de normalização devem ser adotados antes de comparar diferentes amostras. RPKM e TPM normalizam o fator mais importante, sequenciando a profundidade. TMM, DESeq e UpperQuartile podem ignorar características altamente variáveis e / ou altamente expressas., Outros fatores que interferem com comparações intra-amostra envolvem comprimento transcrito, vieses posicionais na cobertura, tamanho médio dos fragmentos e conteúdo GC, que podem ser normalizadas por ferramentas, tais como DESeq, edgeR, baySeq, e NOISeq. Os efeitos do lote podem ainda estar presentes após a normalização, que pode ser minimizado por design experimental apropriado, ou removido por métodos como combate ou ARSyN.
Tabela 5. As ferramentas de normalização para testes de expressão diferencial.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Múltiplas ferramentas bioinformáticas foram desenvolvidas para detectar a partir de dados experimentais. A comparação destas ferramentas de detecção utilizando dados RNA-seq foi realizada pela Ding em 2017, e os resultados são mostrados na Tabela 7. Eles demonstraram que TopHat e sua ferramenta downstream, FineSplice, são as ferramentas mais rápidas, enquanto a PASTA é o programa mais lento. Além disso, AltEventFinder pode detectar o maior número de junções, e RSR detecta o menor número de junções. Outras ferramentas, como o TopHat, são susceptíveis de detectar falsos positivos., Das duas ferramentas que detectam isoformas Articuladas diferentemente, o rMATS é mais rápido do que o rSeqDiff, mas detecta isoformas com menor variação do que o rSeqDiff.
Tabela 7. Detectados como tipos ou isoformas diferentemente Articuladas destas ferramentas (Ding et al. 2017).,
Visualização
Existem muitas ferramentas de bioinformática para a visualização de RNA-seq dados, incluindo o genoma navegadores, tais como ReadXplorer, UCSC navegador, Integrative Genomics Viewer (IGV), Genoma Mapas, Savant, ferramentas especificamente concebidas para o RNA-seq dados, tais como RNAseqViewer, bem como alguns pacotes de expressão gênica diferencial de análise que permitem a visualização, tais como DESeq2 e DEXseq em Bioconductor. Pacotes, como CummeRbund e sashimi plots, também foram desenvolvidos para fins exclusivos de visualização.,
Perfil Funcional
A última etapa de um estudo padrão de transcriptomia é geralmente a caracterização das funções moleculares ou vias nas quais estão envolvidos genes de expressão diferente. Ontologia genética, Biocondutor, DAVID, ou Babelomics contêm dados de anotação para a maioria das espécies-modelo, que podem ser usados para a anotação funcional. As for novel transcripts, protein-coding transcripts can be funcionally annotated using orthology with the help of databases such as SwissProt, Pfam, and InterPro., A ontologia genética (GO) permite alguma permutabilidade de informações funcionais entre ortólogos. Blast2GO é uma ferramenta popular que permite a anotação massiva de transcriptoma completo contra uma variedade de bases de dados e vocabulários controlados. O banco de dados Rfam contém a maioria das famílias de RNA bem caracterizadas que podem ser usadas para a anotação funcional de longas RNAs não codificadoras.a análise avançada do RNA-seq geralmente inclui outros RNA-seq e integração com outras tecnologias, o que é descrito na Figura 4., Mais informações sobre as aplicações de RNA-seq, por favor Veja este artigo aplicações de RNA-Seq.
Figura 3. A análise avançada dos dados RNA-seq.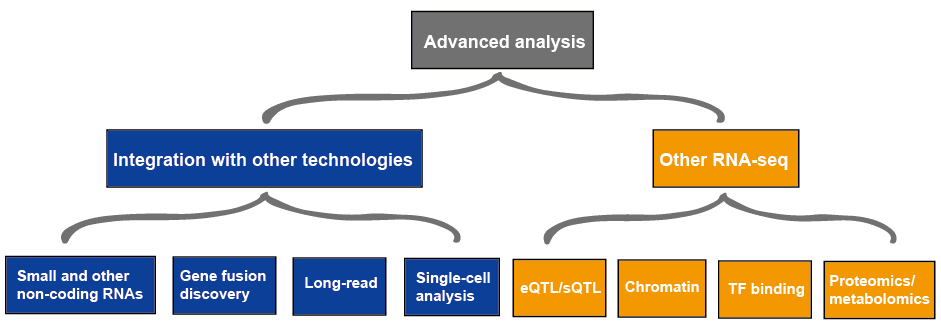
nossos cientistas experientes de Bioinformática são habilidosos em utilizar as ferramentas avançadas de bioinformática para lidar com as numerosas sequências geradas pela sequenciação da próxima e terceira geração. Fornecemos serviços de sequenciamento e bioinformática para genômica, transcriptômica, epigenômica, genômica microbiana, sequenciação de células únicas e sequenciamento PacBio SMRT.