no mês Passado, fui convidado para falar dos Centros para Mendeliana Genômica (CMG) Análise e Desenvolvimento de Métodos de reunião sobre “População, baseado na estimativa de a penetrância em doenças raras”. Aqui está a versão post do blog da minha palestra.o que é a penetração e por que nos importamos?
Penetrance é a probabilidade de desenvolver uma doença específica, dado um genótipo específico., Pode-se falar de penetração dependente da idade, então a porcentagem de pessoas com o genótipo desenvolvendo a doença aos 40 anos, aos 50 anos, e assim por diante; eu geralmente falo em termos de risco de vida, significando a probabilidade de que você alguma vez desenvolver a doença antes de morrer. Inerente a isto é que, para as doenças de adultos, o risco de vida nunca pode ser 100%, porque você sempre pode morrer de outra coisa antes.
Penetrance é extremamente importante para os indivíduos que estão passando por testes genéticos preditivos — a primeira pergunta de muitas pessoas é: “isso significa que eu definitivamente vou ter a doença?”., No entanto, é muitas vezes muito difícil chegar por uma estimativa firme de penetração.métodos tradicionais para estimar a penetração em um mundo ideal, a maneira certa de estimar a penetração seria determinar, desde o nascimento, uma grande coorte de pessoas com um genótipo particular, segui-los até que todos tenham morrido de alguma coisa ou outra, e então perguntar quantos já desenvolveram a doença antes de morrer. Desde que a tecnologia de genotipagem foi inventada há menos de uma vida humana, isso nunca foi feito para qualquer doença.em vez disso, os investigadores usam frequentemente métodos familiares para estimar a penetração., Um estudo típico iria olhar para todos os que foram observados com o genótipo dado, e perguntar quantos têm diease, ou quantos têm doença em uma determinada idade. Os métodos baseados na família sofrem de um viés determinador pervasivo ., apurado sobre a base de apresentar a doença
Como exemplo deste último ponto, em genética de doenças priônicas, apenas 23% de risco as pessoas a escolher a testes genéticos preditivos , e no pedigree de dados a que tive acesso, sabíamos que os genótipos de apenas 22% das pessoas em risco .,todos os fatores listados acima trabalham na mesma direção, tendendo a inflacionar a estimativa de penetração.os investigadores estão cientes destes problemas há muito tempo e propuseram algumas soluções. Como um exemplo, o método de coorte de parentesco envolve a determinação de indivíduos saudáveis aleatoriamente a partir de uma população, genotipando-os, tomando uma história familiar, e comparando curvas de sobrevivência de seus parentes de primeiro grau., Esta é uma solução muito inteligente, mas depende de ser capaz de determinar um número suficiente de pessoas com um genótipo causador de doenças sem determinar a presença de doença. Então ele trabalhou para as variantes BRCA1 e BRCA2 em judeus Ashkenazi americanos, mas para muitas condições genéticas mais raras, é impraticável, porque você precisaria recrutar dezenas ou centenas de milhares de pessoas para encontrar mesmo um indivíduo com um genótipo de interesse.,
métodos baseados na população
por todas as razões descritas acima, é muito útil ter métodos ortogonais baseados na população, para fazer perguntas sobre a penetração. O primeiro insight chave aqui é que uma variante genética completamente penetrante não deve ser mais comum na população do que a doença que causa. Aplicar esta lógica na prática significa que você precisa de boas estimativas de frequência alélica mesmo para variantes incomuns, e isso tem sido difícil de encontrar até recentemente. Exact, um banco de dados de variação genética em 60,706 exomas humanos, oferece novas oportunidades ., Muitos indivíduos foram identificados como casos ou controles para várias doenças complexas comuns, mas nenhum foi determinado para a doença Mendeliana, então exact é uma boa base de dados de referência para o estudo da maioria das doenças genéticas.ao fornecer informações de frequência alélica na população em geral, ExAC, como bases de dados de referência anteriores, como a ESP, deixou claro que a genética clínica tem um grande problema: muitas variantes relatadas como causadoras de doenças genéticas não causam realmente doenças genéticas, ou pelo menos não na maioria das vezes.,
duas bases de dados — HGMD e ClinVar — coletam afirmações da literatura e de laboratórios clínicos afirmando que uma variante genética específica causa uma doença genética particular. Na última contagem, havia mais de 100.000 variantes genéticas causadoras de doenças reportedy únicas nestas bases de dados. A pessoa média na exat tem 54 deles . Obviamente, a pessoa média não tem 54 doenças genéticas., É claro, muito desse excesso é causado por um pequeno número de variantes de alta frequência que obviamente não causam nenhuma doença genética, e grande parte dela pode ser supostamente variantes recessivas encontradas em um estado heterozigótico em exatidão. Mas, mesmo se apenas olharmos para variantes de uma doença dominante genes em um alelo frequência de <1%, ainda vemos 0.89 supostamente patogênicos variantes por pessoa , e isso claramente não é o caso que ~90% das pessoas têm uma doença genética dominante., Então, através do espectro de frequência do alelo, há um monte de variantes supostamente patogênicas que não são tão patogênicas. Quando Anne O’Donnell e eu olhamos para as variantes supostamente patogênicas com as mais altas freqüências de alelos na exat, e perguntamos como elas conseguiram ser mal classificadas como patogênicas, descobrimos que na maioria das vezes o problema remonta a um artigo na literatura que tinha feito uma alegação de patogenia com base em evidências insuficientes.
acima: figuras 3C e 3D de ., Em todo o espectro de frequência do alelo e em ambos os genes da doença dominante e recessiva, há um monte de variantes supostamente patogênicas que aparecem na exat. De alta freqüência (>1%) variantes supostamente patogênicas, alguns são genuinamente patogênicos, alguns são genuinamente associados, mas o traço é benigno, e alguns são erros de anotação em bases de dados — mas a maioria é baseada na literatura com evidências insuficientes.,
Alelo informações de freqüência ExAC tem agora habilitado a mais de 200 variantes genéticas para ser reclassificados do patogênicos para benigno, provavelmente benigna, ou de significado incerto . Estes tipos de reclassificações às vezes desencadeiam um recuo dos autores originais que propuseram que uma variante causa uma doença genética, que pode argumentar que uma variante ainda pode ser patogênica, mas com penetração incompleta. Mas quão incompleta pode ser a penetração incompleta?, Temos de ser quantitativos, porque se o risco ao longo da vida é, no máximo, de 1%, então ainda é razoável dizer que uma variante “causa” uma doença genética, ou é “patogénica”? Enquanto o alelo frequência a informação nunca pode provar que uma variante não tem nenhuma associação para a doença, ele pode colocar limites em que a penetrância pode ser, e em muitos casos, mesmo relativamente raras variantes, é possível mostrar que não há nenhuma maneira uma variante confere um nível de risco em qualquer lugar remotamente perto de 100%.,
para obter quantitativo, precisamos estender a nossa observação anterior-que uma variante genética completamente penetrante não deve ser mais comum na população do que a doença que causa. Isto é tudo matemática simples e genética populacional, mas muitas vezes não é aplicado na prática. Aqui estão duas maneiras que podemos pensar sobre a frequência do alelo ao fazer inferências sobre patogenia e penetração.
frequência alélica máxima credível
diz que está a estudar o Exoma de um doente com doença Mendeliana e a tentar identificar a variante causal., O meu colega James Ware concebeu uma estratégia para filtrar esse exome contra a informação de frequência allele na exat, aproveitando a seguinte lógica., O máximo alelo frequência que é plausível para uma variante para causar uma doença genética dominante é igual a prevalência da doença vezes a heterogeneidade alélica (proporção de casos atribuíveis a uma variante), dividido pelo a penetrância (menos penetrante variantes pode ser mais comum), dividido por 2 (porque nós somos diplóides):
\
Por exemplo, o prion doença faz com que em 1 em 5.000 as mortes, e a variante mais comum (E200K) é encontrada em 5% dos casos , a 100% penetrante variante não pode ter alelo frequência maior de 0,0005% (1 em cada 200.000 mil) ., Cardiomiopatia afeta 1 em cada 500 pessoas, a variante mais comum é encontrada em <2% dos casos, então uma variante penetrante de 50% não pode ter uma frequência alélica superior a 0,004% . A fórmula para as doenças recessivas é um pouco mais complicada, mas James também tem trabalhado para fora e é descrito em .
Então, considerando que, historicamente, as pessoas têm muitas vezes filtrado variantes com um alelo frequência >0.1% quando tentar identificar a causa de uma doença dominante , podemos ser muito mais rigorosos., A ressalva é que em baixas contagens de alelos, a nossa capacidade de estimar a frequência do alelo é limitada pela variância de amostragem. Por exemplo, se olharmos para as variantes observadas a uma frequência alélica de 1% entre os europeus em ESP, estas variantes também têm uma frequência de cerca de 1% entre os europeus exactos. Mas as variantes com uma frequência de 0,1% em ESP tendem a ser ligeiramente mais raras em exatidão, e a maioria dos singletons (variantes vistas exatamente uma vez em ESP) não reaparecem uma segunda vez em exatidão.
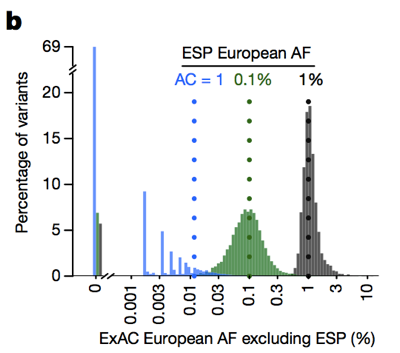
acima: figura 3B de . Quanto menor a contagem de alelos, menos boa é a estimativa da frequência de alelos que fornece.,
portanto, quanto menor a contagem de alelos, mais conservador precisamos ser. Nós concebemos uma estrutura para fazer isso usando o limite superior de 95% da distribuição de Poisson em quantos alelos podem ser observados em uma dada frequência, e temos valores pré-computados para todos os exatos (disponíveis em FTP) que você pode usar — leia mais sobre os métodos em . James também criou este aplicativo Web útil que lhe permite explorar o que a “frequência alélica máxima credível” deve ser para a sua doença de interesse.,
Inherent in this approach is that the lower the penetrance of a variant, the higher frequency it might have in the general population. Mas você também tem que descobrir que se a penetração é bastante baixa, digamos, menos de 10%, então a utilidade clínica dessa variante também é baixa. James e Nicky Whiffin apresentaram dados para mostrar que quase toda a utilidade clínica da sequenciação em cardiomiopatia vem de variantes com uma frequência de <0,001% — variantes mais comuns cumulativamente contribuem pouco, se algum, risco .,estimation and bounds of lifetime risk
Remember that penetrance is the probability of disease given a particular genótipo. Ou, se considerarmos um modelo alélico ao invés de genotípico, a probabilidade de doença dada um alelo particular. Podemos escrever isto como P (D|A). Uma vez que o façamos, torna-se claro que, pelo teorema de Bayes,
\
cada um destes Termos tem um significado particular:
Note aqui que “controle populacional” significa um grupo não selecionado para a presença, nem para a ausência, da doença. Apenas uma parte da população em geral.
So:
\
This logic is nothing new., O uso do teorema de Bayes para estimar o risco de doença remonta , pelo menos, à estimativa do risco de câncer em fumantes, e sua aplicação à genética tem sido considerada por quase tanto tempo . Mas para que esta equação funcione para as doenças raras, você precisa de estimativas bastante boas de frequência de alelos de controle de casos e população, e essas têm sido difíceis de encontrar até recentemente. Portanto, graças à exat, há um número crescente de situações em que esta equação é relevante.
Aqui está o código R que escrevi (originalmente aqui) para estimar a penetração com base nesta fórmula.,
Se você não quiser executar o código R você mesmo, James Ware implementou-o na página “penetrance” deste aplicativo web para que você possa simplesmente ligar seus números em seu navegador.a fim de estimar intervalos de confiança de 95% na penetração, eu adotei a abordagem de . Entrada o alelo de contagem (AC) e o número de indivíduos (N) para casos e controles, e o limite superior do IC de 95%, é calculado com base no superior CI de 95% da distribuição binomial para o caso alelo frequência e inferior do IC 95% para os controles., Inversamente, o limite inferior da penetração é baseado no limite inferior da frequência do alelo de caso e no limite superior da frequência do alelo de controlo. Pode-se legitimamente questionar que, como esta fórmula utiliza 95% CIs em ambos os valores de frequência alelos, os intervalos de confiança resultantes são maiores do que deveriam ser. Você também pode, com razão, tergiversar que a distribuição binomial não é um bom estimador de baixa alelo conta, devido ao viés ilustrado na Figura 3B mostrado acima (e eu certamente nunca iria aplicar esta fórmula para gestações únicas variantes observadas apenas uma vez em ExAC)., Mas no final do dia, por razões que vou discutir mais perto do fim deste post, esta fórmula é realmente melhor usada para obter um ballpark, ordem de magnitude estimativa de penetração. Se você está procurando uma estimativa de ponto de penetração extremamente precisa, esta abordagem provavelmente não vai funcionar para você de qualquer maneira.se reorganizar a equação, outra forma de pensar é:
\
isto significa que o risco aumentado entre as pessoas com um genótipo é proporcional à razão entre a frequência do alelo de controlo de casos e da população., Assim, uma variante que aumenta o risco em 200 vezes deve ser 200 vezes mais comum entre os casos do que na população em geral. (Note que esta razão de frequências alelas é ligeiramente diferente da razão de probabilidades, embora as duas medidas convergam para variantes muito raras.)
application to prion disease
andámos através desta lógica num estudo que publicámos no início deste ano, quantificando a penetração das variantes da doença de prião ., Preocupo — me com a doença de prião por uma razão pessoal — a minha mulher tem uma variante patogênica na PRNP-mas acontece que a doença de prião também é um grande caso de teste para usar a lógica acima para estimar a penetração. Nenhum dos indivíduos em exact v1 foram apurados sobre a doença neurodegenerativa, então exat realmente é um bom conjunto de dados de controle populacional para a doença de prion. E como as doenças prion são “notificáveis”, os Centros Nacionais de vigilância têm casos excepcionalmente bons para determinar, e graças à sua generosidade em compartilhar dados, fomos capazes de acumular um conjunto de dados de 10.460 casos sequenciados.,
descobrimos que as variantes > 60 notificadas como causadoras da doença de prião cumulativamente têm 52 alelos exactamente. Isso significa que quase 1 em cada 1000 pessoas tem uma dessas variantes, e assim, estas variantes são cumulativamente muito mais comuns do que toda a doença de prião (que causa ~1 em cada 5.000 mortes), muito menos toda a doença de prião genético (apenas ~15% dos casos são genéticos). Isto é suficiente para nos dizer que nem todas estas variantes podem ser totalmente penetrantes. Para determinar quais variantes foram os culpados, comparamos com a série de casos., Variantes com excelentes evidências prévias de patogenicidade (modelos mendelianos de segregação e mouse) eram comuns em casos e ausentes de exat, consistentes com a penetração completa ou quase completa. A maior parte da contagem de alelos em excesso na exat foi contribuída por variantes que eram pouco comuns em casos e tinham fraca evidência prévia de patogenicidade — estas variantes são provavelmente benignas ou contribuem apenas com um baixo risco. Pelo menos três variantes pareceram intermediárias, pois eram muito comuns em controles para Penetração total, mas ainda enriquecidas em casos sobre controles.,
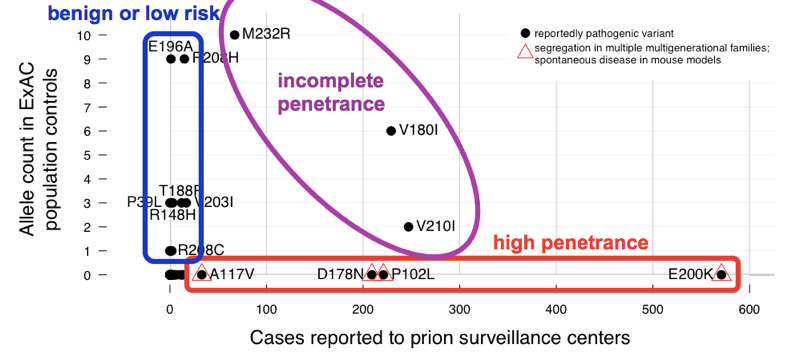
Acima: uma versão anotada da Figura 2 .
Quando estimamos a penetração para cada variante, usando a fórmula P(D|A) acima, descobrimos que existe um espectro inteiro de penetração para variantes PRNP.
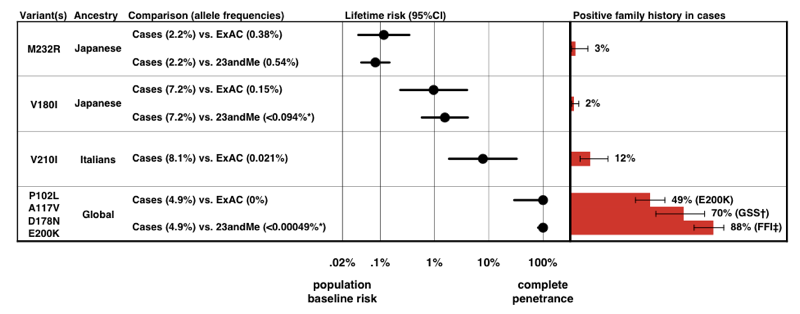
Acima: Figura 3 .
Note a escala no eixo x — para uma doença tão rara que a probabilidade prévia de desenvolvimento é de apenas 0, 02%, mesmo um aumento de 50 vezes no risco é apenas 1% de risco ao longo da vida., Tranquilamente, as estimativas de penetração que derivamos da informação de frequência alélica por si só estão muito bem de acordo com a proporção de casos que apresentam uma história familiar positiva.este trabalho já levou a uma mudança de prognóstico para alguns indivíduos que originalmente tinham sido aconselhados de que estavam em risco de variantes de alta penetração-veja e Erika Check Hayden artigo sobre exat. Podes ler a viagem pessoal da minha e da Sonia com este estudo aqui.,
A aplicação à NR1H3
esclerose múltipla (MS) é uma doença complexa com muitos factores de risco genéticos , mas não se sabe que exista nenhuma forma Mendeliana da doença. No início deste ano, um estudo relatou que uma variante missense em um receptor de hormônio nuclear — NR1H3 R415Q — causa a primeira forma Mendeliana de MS . Esta alegação foi baseada na segregação dominante com a doença em duas famílias, mas a pontuação do LOD foi apenas 2,2 — abaixo do limiar para o significado do genoma em estudos de ligação familiar, que é mais como 3.0 ou 3.6 . E a variante em questão tem uma frequência alélica de 0.,031% nos Europeus não finlandeses. Isso pode não parecer uma frequência alélica alta, mas acaba por ser muito alta para que esta variante cause esclerose Mendeliana .considera que a em tem um risco ao longo da vida (na população em geral) de 0, 25% nas mulheres e 0, 14% nos homens . Se 0.06% de pessoas na população em geral são R415Q heterozigotos, e se nem a metade delas passou a desenvolver MS, em seguida, esta variante só conta 0,03% da população, o desenvolvimento de MS. Então, se um total de 0,25% de pessoas desenvolver MS, em seguida, cerca de 12% deles deve ter esta variante., Em vez disso, a variante foi encontrada apenas em 1 indivíduo de uma série de casos de 2 053 doentes em.
isto funciona para uma frequência alélica de 0,024% nos casos, ou 0,049% se permitirmos que 2 casos sejam contados na série de casos. Isto não é significativamente maior do que a frequência na exat. Mas se esta variante causa Em, deve ser mais comum em casos-muito mais comum. Lembre-se da nossa fórmula reorganizada anteriormente: P(D|A)/P(D) = P(A|D)/P(a). Isto significa que se uma variante aumenta o risco em X-vezes, deve ser X vezes mais comum nos controles. Por isso, se o risco inicial de EM for 0.,25% e esta variante é 50% penetrante, deve ser 50/.25 = 200 vezes mais comum em casos do que controles. Se ele mesmo tinha 10% de penetração, ele ainda deve ser 10/.25 = 40 vezes mais comum em casos do que em controles. Alternativamente, você pode pensar em termos de rácios de probabilidades em vez de probabilidades. O risco ao longo da vida de 0,25% na população em geral significa 1:399 probabilidades, e se R415Q conferiu 50% de risco ao longo da vida, isso seria 50:50 probabilidades. (50/50)/(1/399) = 399, então a razão de probabilidade para R415Q teria que ser 399 para que esta variante tivesse 50% de penetração.,
em vez disso, se aplicarmos a nossa fórmula usando o código R de antes, assumindo 0.25% de risco de base e baseando o cálculo em 2 alelos em 2.053 casos, versus 21 alelos em 33.369 indivíduos exactos, descobrimos que o limite superior do IC de 95% na penetração é de 2,2%. Assim, mesmo que o R415Q estivesse associado ao risco de MS, não poderia conferir mais de 2,2% de risco ao longo da vida de desenvolvimento de MS .,
em sua resposta formal e em PubMed Commons, os autores levantaram uma comparação com LRRK2 G2019S na doença de Parkinson, que todos concordam ser patogênica, mas que também é encontrada na exat e tem apenas uma pequena proporção de probabilidades, estimada em 9,6 . Para essa variante, a matemática funciona. A doença de Parkinson é, pelo menos, uma ordem de magnitude mais prevalente do que a em, com o risco de vida estimado em qualquer lugar de 3,7% a 6,7% . Esta ordem de magnitude de prevalência maior significa que o enriquecimento de ~10 vezes observado-LRRK2 G2019S é encontrado em aproximadamente 0.,1% dos controles e 1% dos casos — é aproximadamente consistente com o relatado risco de vida de ~32% de Parkinson conferido por esta variante . Estes detalhes quantitativos importam, e são diferentes para cada variante e cada doença. É por isso que as fórmulas discutidas neste post são úteis, apesar de apenas fornecerem estimativas muito aproximadas e estarem sujeitas a várias advertências, como explicado abaixo.
caveats
em ambas as aplicações descritas acima, a informação de frequência do alelo foi usada para obter uma estimativa aproximada da penetração., Na doença de prião, pudemos mostrar que variantes anteriormente presumidas altamente penetrantes conferiam maior risco de vida na ordem de 0,1%, 1%, ou 10%. Na história NR1H3, a informação de frequência alélica foi suficiente para mostrar que a variante supostamente causal não poderia conferir mais do que alguns por cento de risco de vida.
mas tentar usar dados de frequência alélica para obter uma estimativa mais apertada de penetração seria muito desafiador. Por exemplo, estudos baseados na família discordaram sobre a penetração do PRNP E200K, com estimativas que variam de 60% a 90% de risco ao longo da vida ., Desde que o estudo prion saiu, algumas pessoas de famílias E200K perguntaram-me se os dados exactos podem ajudar a reduzir onde o risco está dentro deste intervalo. A resposta é, infelizmente, ele não pode.
Aqui estão as razões mais importantes que eu acho que toda a penetrância estimativas baseadas em alelo frequência devem ser tomadas com um grão de sal:
- Se uma variante é altamente penetrante, é difícil obter uma série de casos que não contêm relacionados indivíduos. Se sua série de casos tem relações, então tecnicamente você não tem uma estimativa imparcial de P (A|D).,se uma doença é fatal, então é difícil obter uma série de controle populacional que não é pelo menos um pouco esgotada de pessoas com variantes que causam essa doença. Então você também não tem uma estimativa imparcial de P(a).as comparações da frequência dos alelos de caso e controlo são vulneráveis à confusão por estratificação populacional. No estudo prion, nós não tínhamos dados de SNP em todo o genoma sobre casos, então não havia maneira de controlar perfeitamente para isso.,muitas variantes causais de doenças raras são tão raras que mesmo com exat, ainda não temos estimativas suficientemente precisas da frequência dos alelos para dar uma resposta melhor do que uma resposta aproximada.
Com tudo o que disse, de base populacional alelo frequência estimativa ainda é uma boa maneira de ficar difícil de ordem de magnitude das estimativas de a penetrância e para executar verificações de sanidade se uma variante genética poderia plausivelmente ser causal de uma doença rara.