Siste måned var jeg invitert til å snakke på Sentrene for Mendelian Genomics (CMG) Analyse og Metoder for Utvikling møtet om «Befolkningen-basert estimering av penetrance i sjeldne sykdommen». Her er blogginnlegget versjon av mitt foredrag.
hva er penetrance og hvorfor vi bryr oss?
Penetrance er sannsynligheten for å utvikle en bestemt sykdom gitt en bestemt genotype., Man kan snakke om alder-avhengige penetrance, slik at andelen av mennesker med genotype å utvikle sykdommen ved fylte 40 år, etter alder 50, og så videre; jeg snakker vanligvis i form av levetid risiko, noe som betyr at sannsynligheten for at du stadig utvikle sykdommen før du dør. Som ligger i dette er at, for voksne-utbruddet sykdommer, levetid risikoen kan aldri helt 100%, fordi du alltid kan dø av noe annet først.
Penetrance er enormt viktig for personer som gjennomgår prediktiv genetisk testing — for mange mennesker er første spørsmålet er, «betyr det at jeg kommer definitivt til å få sykdommen?»., Men det er ofte svært vanskelig å komme med en fast estimat av penetrance.
tradisjonelle metoder for å estimere penetrance
I en ideell verden, den riktige måten å anslå penetrance ville være å bringe på det rene, fra fødselen, en stor kohort av personer med en bestemt genotype, følge dem helt til alle har dødd av noe eller andre, og spør deretter om hvor mange som noensinne er utviklet sykdommen før de døde. Siden genotyping teknologi ble oppfunnet mindre enn ett menneske som levde siden, dette har aldri blitt gjort for noen sykdom.
i Stedet, forskere bruker ofte familie-baserte metoder for å beregne penetrance., En typisk studien vil se på alle som har blitt observert med en gitt genotype, og spør hvor mange har diease, eller hvor mange har sykdommen ved en viss alder. Familie-baserte metoder lider av gjennomgripende ascertainment bias ., fastsatt på grunnlag av å presentere med sykdom
Som et eksempel på dette siste punktet, i genetisk prion sykdom, bare 23% på utsatte mennesker velger prediktiv genetisk testing , og stamtavle data som jeg har hatt tilgang til, vi visste genotypes av bare 22% av utsatte individer .,
Alle av de faktorer som er nevnt ovenfor jobbe i samme retning, har en tendens til å blåse opp en beregning av penetrance.
Forskere har vært klar over disse problemene i lang tid, og har foreslått noen løsninger. Som ett eksempel, den pårørende-kohorten metoden innebærer å bringe på det rene friske individer tilfeldig fra en populasjon, genotyping dem, å ta en familie historie, og sammenligne overlevelse kurver av deres første-graders slektninger., Dette er en veldig smart løsning, men det er avhengig av å være i stand til å fastslå et stort nok antall av mennesker med en sykdom som forårsaker genotype uten å bringe på det rene på forekomsten av sykdommen. Så det fungerte for BRCA1 og BRCA2 varianter i Amerikansk Ashkenazi Jøder , men for mange sjeldne genetiske forhold, er det upraktisk, fordi du vil trenge for å rekruttere flere titalls eller hundrevis av tusenvis av mennesker til å finne enda en person med en genotype av interesse.,
befolkningen-baserte metoder
For alle de grunner som er beskrevet ovenfor, er det svært nyttig å ha ortogonale, befolkning-baserte metoder, for å stille spørsmål om penetrance. Den første nøkkelen innsikt her er at en helt penetrant genetisk variant bør ikke være mer vanlig i befolkningen enn den sykdom som forårsaker det. Søker denne logikk i praksis betyr det at du må ha gode estimater av allelet frekvens selv for mindre vanlige varianter, og det er blitt vanskelig å komme med inntil nylig. ExAC, en database med genetiske variasjonen i 60,706 menneskelige exomes, gir nye muligheter ., Mange personer i ExAC ble konstatert som saker eller kontrollerer for ulike felles, komplekse sykdommer, men ingen ble konstatert for Mendelian sykdom, så ExAC er en god referanse-database for å studere de fleste genetiske sykdommer.
Ved å gi allelet frekvens informasjon i den generelle befolkningen, ExAC, som tidligere referanse databaser som ESP , har gjort det klart at klinisk genetikk har et stort problem: mange varianter rapportert å forårsake genetiske sykdom ikke faktisk forårsake genetiske sykdommen, eller i det minste det meste av tiden.,
To databaser — HGMD og ClinVar — samle påstander fra litteraturen og fra kliniske laboratorier som sier at en bestemt genetisk variant forårsaker en bestemt genetisk sykdom. Ved siste opptelling var det over 100.000 unike reportedy sykdom som forårsaker genetiske varianter i disse databasene. Den gjennomsnittlige personen i ExAC har 54 av dem . Selvsagt, er den gjennomsnittlige personen ikke faktisk har 54 genetiske sykdommer., Selvfølgelig, mye av dette overflødig er forårsaket av et lite antall av svært høy frekvens varianter som åpenbart ikke vil føre til noen genetisk sykdom, og mye av det kan være angivelig recessiv varianter som blir funnet i en heterozygot staten i ExAC. Men selv om vi bare ser på varianter i dominant sykdom gener på et allel frekvens av <1%, ser vi fortsatt 0.89 angivelig patogene varianter per person , og det er helt klart ikke tilfelle at ~90% av mennesker har en dominerende genetisk sykdom., Så over allelet frekvens spekteret, det er mange av angivelig patogene varianter som ikke er så patogene. Når Anne O ‘ Donnell og jeg så på den angivelig patogene varianter med den høyeste allelet frekvenser i ExAC, og spurte hvordan de hadde klart å bli klassifisert som patogene, fant vi at det meste av tiden problemet spores tilbake til en artikkel i litteratur som hadde laget et krav om pathogenicity basert på utilstrekkelig bevis.
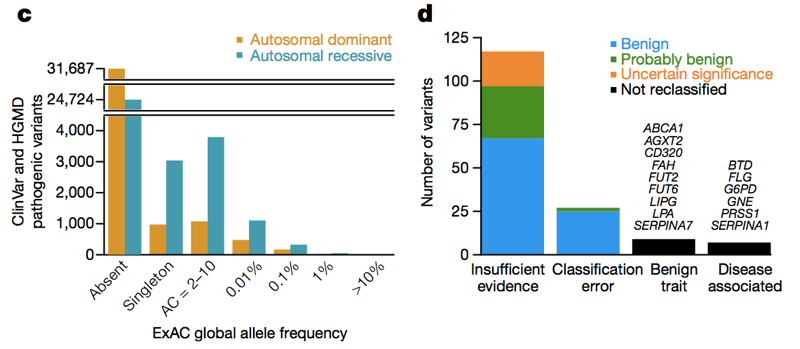
Over: Tall 3C og 3D fra ., Over allelet frekvensspekteret, og i både dominant og recessiv sykdom gener, det er mange av angivelig patogene varianter som vises i ExAC. For høy (>1%) frekvens angivelig patogene varianter, noen er genuint patogene, noen er genuint trekk-forbundet, men trekket er godartet, og noen er feil av kommentarene i databaser, men de fleste er basert på litteratur med utilstrekkelige bevis.,
Allelet frekvens informasjon fra ExAC har nå aktivert over 200 genetiske varianter å bli reklassifisert fra patogene til godartet, sannsynlig benign, eller av usikker betydning . Disse sorterer av reklassifisering noen ganger utløse pushback fra den opprinnelige forfatterne som er foreslått som en variant fører til en genetisk sykdom som kan hevde at en variant kan fortsatt være patogene, men med ufullstendig penetrance. Men akkurat hvor «ufullstendig» kan ufullstendige penetrance være?, Vi trenger å få kvantitative, fordi hvis levetid risiko på det meste 1%, så er det likevel rimelig å si at en variant «fører til» en genetisk sykdom, eller er «patogene»? Mens allelet frekvens informasjon kan aldri bevise at en variant har ingen tilknytning til sykdom, det kan sette grenser på hva det er mulig penetrance kan være, og i mange tilfeller, selv for nokså sjeldne varianter, det er mulig å vise at det er ingen måte en variant gir en risiko hvor som helst eksternt nær 100%.,
for Å få kvantitative, trenger vi å utvide vår tidligere observasjon — som en helt penetrant genetisk variant bør ikke være mer vanlig i befolkningen enn den sykdom som forårsaker det. Dette er enkel matematikk og populasjonsgenetikk, men det er altfor ofte ikke anvendt i praksis. Her er to måter vi kan tenke allelet frekvens når du gjør slutninger om pathogenicity og penetrance.
maksimalt troverdig allelet frekvens
Sier du studerer exome av en pasient med Mendelian sykdom og prøver å identifisere den kausale variant., Min kollega James Ware har utarbeidet en strategi for filtrering exome mot allelet frekvens informasjon i ExAC, å dra nytte av følgende logikk., Maksimal allelet frekvens som er rimelig for en variant for å føre en dominerende genetisk sykdom er lik forekomst av sykdommen ganger genetisk mangfold (andel av tilfellene kan tilskrives en variant) delt på penetrance (mindre penetrant varianter kan være mer vanlig), dividert med 2 (fordi vi er diploid):
\
For eksempel, prion forårsaker sykdommen i 1 5000 dødsfall, og den vanligste varianten (E200K) er funnet i 5% av tilfellene , så en 100% penetrant variant kan ikke muligens har allelet frekvens større enn 0.0005% (1 i 200,000) ., Kardiomyopati påvirker 1 500 personer, den vanligste varianten er funnet i <2% av tilfellene, så en 50% penetrant variant kan ikke ha et allel frekvens større enn 0.004% . Formelen for recessiv sykdommer er ett hakk mer komplisert, men James har også jobbet det ut, og det er beskrevet i .
Så, mens historisk folk har ofte filtrert ut varianter med et allel frekvens >0.1% når du prøver å identifisere årsaken til en dominant sykdom , kan vi faktisk bli mye strengere., Det forbeholdet er at ved lave allelet teller, vår evne til å anslå allelet frekvens er begrenset av sampling-variansen. For eksempel, hvis vi ser på varianter sett på en 1% – allelet frekvens blant Europeerne i ESP, disse variantene har også om lag 1% frekvens blant ExAC Europeere. Men varianter med et 0.1% frekvens i ESP har en tendens til å være litt sjeldnere i ExAC, og de fleste singletons (varianter sett nøyaktig en gang i ESP) ikke igjen en gang i ExAC.
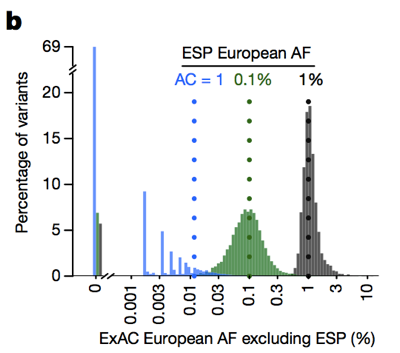
Over: Figur 3B fra . Den lavere allelet telle, de mindre gode en beregning av allelet frekvens det gir.,
Derfor, nedre allelet telle, jo mer konservative vi trenger å være. Vi har utviklet et rammeverk for å gjøre dette ved hjelp av 95% øvre grense av Poisson-fordeling på hvor mange alleler kan observeres på en gitt frekvens, og har pre-beregnede verdier for alle ExAC (tilgjengelig på FTP) som du kan bruke — les mer om metoder i . James har også laget denne hendige web app som lar deg utforske hva «maksimal troverdig allelet frekvens» bør være for din sykdom av interesse.,
Iboende i denne tilnærmingen er at jo lavere penetrance av en variant, jo høyere frekvens det kan ha i den generelle befolkningen. Men du har også å finne at hvis penetrance er ganske lav, si, mindre enn 10%, så den kliniske nytten av at varianten er også lav. James og Nicky Whiffin har presentert data for å vise at nesten alle av klinisk nytte av sekvensering i kardiomyopati kommer fra varianter med en frekvens på <0.001% — mer vanligste variantene kumulativt bidra liten, om noen, risiko .,
estimering og grensene for livet risiko
Husk at penetrance er sannsynligheten for sykdom er det gitt en bestemt genotype. Eller, hvis vi vurdere et allel snarere enn genotypic modell, er sannsynligheten for sykdom gitt et bestemt allel. Vi kan skrive dette som P(D|A). Når vi gjør det, blir det klart at, av Bayes’ teorem,
\
Hver av disse vilkårene, har en spesiell betydning:
Merk her at «befolkningen kontroller» betyr at en gruppe ikke er valgt for tilstedeværelse eller fravær av sykdom. Bare en bit av den generelle befolkningen.
Slik:
\
Denne logikken er ikke noe nytt., Bruk av Bayes’ teorem å anslå sykdom risiko dateres tilbake minst til estimering av risiko for kreft hos røykere , og dens program for å genetikk har vært ansett for nesten like lenge . Men for denne ligningen til å arbeide for sjeldne sykdommer, trenger du ganske gode estimater av saken og befolkning allelet frekvens, og de har vært vanskelig å komme med inntil nylig. Så takk til ExAC, det er et økende antall situasjoner der denne ligningen er relevant.
Her er R-kode jeg har skrevet (opprinnelig her) til å beregne penetrance basert på denne formelen.,
Hvis du ikke ønsker å kjøre på R-koden selv, James Ware har implementert den i «penetrance» fanen på denne web app, slik at du kan bare koble til din tall i nettleseren.
for å beregne 95% konfidensintervall på penetrance, jeg har tatt i bruk tilnærming av . Har du innspill allelet telle (AC) og antall individer (N) for tilfeller og kontroller, og den øvre grense for 95%CI er beregnet basert på den øvre 95%CI av den binomiske fordelingen for saken allelet frekvens og lavere 95%CI for kontroller., Og motsatt, jo lavere er bundet av penetrance er basert på den nedre grense av saken allelet frekvens og den øvre grense for kontroll allelet frekvens. Du kan rette krangle at fordi denne formelen bruker 95%CIs på begge allelet frekvens verdier, som følge av konfidensintervall blir større enn de bør være. Du kan også rette krangle at den binomiske fordelingen ikke er en god estimator ved lave allelet teller, på grunn av fordommer illustrert i Figur 3B vist ovenfor (og jeg ville sikkert aldri bruk denne formelen til å singletons — varianter observert kun en gang i ExAC)., Men på slutten av dagen, av grunner jeg vil diskutere nærmere slutten av dette innlegget, denne oppskriften er egentlig best for å få et ballpark, bestilling-av-anslå omfanget av penetrance. Hvis du leter etter en ekstremt presis punktestimatet av penetrance, hele denne tilnærmingen vil sannsynligvis ikke fungere for deg uansett.
Hvis du vil omorganisere ligningen, en annen måte å tenke på det er:
\
Dette betyr at den økte risikoen blant personer med genotype er proporsjonal med forholdet mellom sak å befolkning allelet frekvens., Så en variant som øker risikoen ved 200-brett skal være 200 ganger mer vanlig blant tilfeller enn det er i befolkningen generelt. (Merk at dette forholdet allelet frekvenser er litt annerledes enn odds ratio selv om de to tiltak konvergerer for svært sjeldne varianter.)
søknad til prion sykdom
Vi gikk gjennom denne logikken i en studie vi har publisert tidligere i år, kvantifisering penetrance av prion sykdom varianter ., Jeg bryr meg om prion sykdom for en personlig grunn — min kone havner en sykdomsfremkallende variant i PRNP — men det viser seg at prion sykdom er også en god test for å bruke de ovennevnte logikk for å anslå penetrance. Ingen av individene i ExAC v1 ble konstatert på nevrodegenerativ sykdom, så ExAC virkelig er en god bestand kontroll dataset for prion sykdom. Og fordi prion sykdommer er «meldepliktig», nasjonal overvåking sentre har usedvanlig god sak ascertainment, og takket være deres raushet i å dele data, vi var i stand til å samle et dataset av 10,460 sekvensert tilfeller.,
Vi har funnet at >60 varianter rapportert å forårsake prion sykdom kumulativt har 52 alleler i ExAC. Det betyr at nesten 1 av 1000 mennesker har en av disse variantene, og dermed er disse variantene er kumulativt mye mer vanlig enn alle prion sykdom (som fører til ~1 i hver 5 000 dødsfall), la alene all genetisk prion sykdom (bare ~15% av tilfellene er genetisk). Dette er nok til å fortelle oss at ikke alle av disse variantene kan muligens være fullt penetrant. For å finne ut hvilke varianter var den skyldige, vi i forhold til saken serien., Varianter med utmerket før bevis for pathogenicity (Mendelian segregering og mus modeller) var vanlig i de tilfeller og fraværende fra ExAC, i samsvar med fullstendig eller nesten fullstendig penetrance. De fleste av de overskytende allelet teller i ExAC var bidratt med varianter som var uvanlig i saker og hadde en svak før bevis for pathogenicity — disse variantene er trolig godartet eller bare vil bidra med en lav risiko. Minst tre varianter dukket opp mellomliggende, som de var også vanlig i kontroller for full penetrance, men fortsatt beriket i tilfeller over kontroller.,
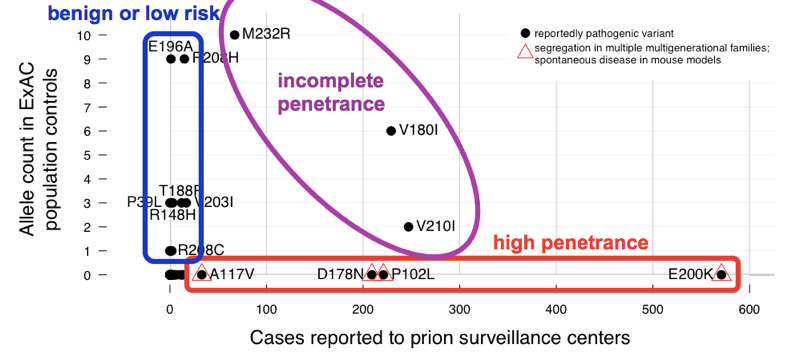
Over: en kommentert versjon av Figur 2 fra .
Når vi har beregnet penetrance for hver variant, ved å bruke P(D|A) formelen ovenfor, finner vi at det er et stort spekter av penetrance for PRNP-varianter.
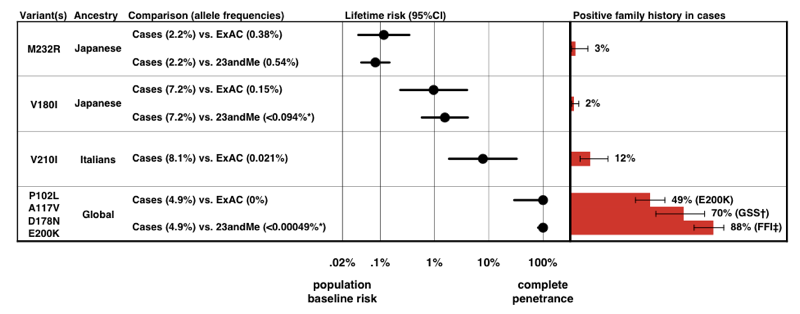
Over: Figur 3 fra .
Merk skalaen på x-aksen — for en sykdom som er så sjeldne at de før sannsynligheten for å utvikle det er bare 0.02%, selv en 50-fold økning i risikoen er bare 1% levetid risiko., Betryggende, det penetrance anslår at vi stammer fra allelet frekvens informasjon alene accord ganske godt med den andel av tilfellene som er tilstede med en positiv familiehistorie.
Dette arbeidet har allerede ført til en endring i prognosen for noen personer som hadde opprinnelig vært sagt at de var i fare for høy-penetrance varianter — se og Erika Sjekk Hayden ‘ s artikkel om ExAC. Du kan lese min og Sonia ‘ s personlige reise med denne studien her.,
søknad til NR1H3
Multippel sklerose (MS) er en kompleks sykdom med mange genetiske risikofaktorer , men ingen Mendelian formen av sykdommen er kjent for å eksistere. Tidligere dette året, en studie rapporterte at en missense-variant i en kjernefysisk hormon-reseptor — NR1H3 R415Q — forårsaker den første noensinne Mendelian form av MS . Denne påstanden var basert på dominerende segregering med sykdom i to familier, men LOD score var det kun 2,2 — under terskelen for genome-wide betydning i familien sammenhengen studier, som er mer lik 3.0 eller 3.6 . Og den varianten i spørsmålet har et allel frekvens av 0.,031% i ExAC ikke-finsk Europeere. Som kanskje ikke høres ut som en høy allelet frekvens, men det viser seg å være altfor høy for denne varianten til å føre Mendelian MS .
Tenk at MS har en levetid risiko (i befolkningen) på 0,25% hos kvinner og 0.14% hos menn . Hvis 0.06% av personer i den generelle befolkningen er R415Q heterozygote katter, og selv om halvparten av dem som gikk på å utvikle MS, så denne varianten alene ville konto for 0,03% av befolkningen å utvikle MS. Så hvis totalt 0,25% av mennesker å utvikle MS, da ca 12% av dem som skal ha denne varianten., I stedet, den varianten ble bare funnet i 1 person ut av en sak rekke 2,053 MS-pasienter .
Dette virker ut til et allel frekvens av 0.024% i tilfeller, eller 0.049% hvis vi tillater for 2 tilfeller å bli regnet med i de tilfelle serien. Dette er ikke vesentlig høyere enn frekvensen i ExAC. Men hvis denne varianten fører til MS, bør det være mer vanlig i tilfeller — er mye mer vanlig. Husk vår omorganisert tidligere formel: P(D|A)/P(D) = P(A|D)/P(A). Dette betyr at hvis en variant øker risikoen ved X-kast, skal det være X ganger mer vanlig hos kontrollene. Så hvis baseline risiko for MS er 0.,25%, og denne varianten er 50% penetrant, bør det være 50/.25 = 200 ganger mer vanlig i tilfeller enn kontroller. Hvis det hadde 10% penetrance, bør det fortsatt være 10/.25 = 40 ganger mer vanlig i tilfeller enn i kontrollene. Alternativt, kan du tenke i form av odds ratio i stedet for sannsynligheter. 0,25% levetid risiko i befolkningen generelt betyr 1:399 odds, og hvis R415Q gitt 50% levetid risiko, som ville være 50:50 odds. (50/50)/(1/399) = 399, så odds ratio for R415Q ville ha til å være 399 for denne varianten til å ha 50% penetrance.,
i Stedet, hvis vi anvender vår formel ved å bruke R-kode fra tidligere, forutsatt 0.25% baseline risiko og basere beregningen på 2 alleler på 2,053 tilfeller, mot 21 alleler i 33,369 ExAC personer, finner vi at den øvre grense for 95% CI på penetrance er på 2,2%. Så selv om R415Q var knyttet til MS risiko, det kunne ikke gi mer enn 2,2% levetid risiko for å utvikle MS .,
I sitt formelle tilsvar og i PubMed Commons forfatterne reist en sammenligning til LRRK2 G2019S i Parkinson ‘ s sykdom, som alle er enige om er patogene, men som også er funnet i ExAC og har bare en beskjeden odds ratio, anslått til 9.6 . For at varianten, matematikk fungerer ut. Parkinsons sykdom er minst en størrelsesorden mer utbredt enn MS, med levetid risiko estimert hvor som helst fra 3,7% til 6.7% . Denne størrelsesorden større utbredelse betyr at ~10-brett berikelse som har blitt observert — LRRK2 G2019S er funnet i omtrent 0.,1% av kontrollene og 1% av tilfellene — er omtrent i samsvar med de rapporterte ~32% levetid risiko for Parkinson ‘ s tildelt av denne varianten . Disse kvantitative opplysninger saken, og er forskjellig for hver variant og hver sykdom. Det er derfor formler som er diskutert i dette innlegget er nyttig, selv om de bare gir svært grove anslag og er underlagt flere begrensninger, som forklart nedenfor.
begrensninger
I begge programmene som er beskrevet ovenfor, allelet frekvens informasjonen ble brukt til å få et grovt anslag på penetrance., I prion sykdom, vi var i stand til å vise at varianter tidligere antatt svært penetrant gitt levetid risiko mer på rekkefølgen på 0,1%, 1%, eller 10%. I NR1H3 historie, allelet frekvens informasjon var tilstrekkelig til å vise at de angivelig årsakssammenheng variant kunne ikke gi mer enn et par prosent levetid risiko.
Men prøver å bruke allelet frekvens data for å få en strammere estimat av penetrance ville være svært utfordrende. For eksempel, familie-baserte studier har ikke kommet til enighet på penetrance av PRNP E200K, med anslag som varierer fra 60% til 90% levetid risiko ., Siden prion studie kom ut, og jeg har hatt et par folk fra E200K familier spør meg om ExAC data kan bidra til å begrense hvor risikoen er innenfor dette området. Svaret er, dessverre, det kan det ikke.
Her er de viktigste grunnene til hvorfor jeg tror alle penetrance estimater basert på allelet frekvens må tas med en klype salt:
- Hvis en variant er svært penetrant, så det er vanskelig å få tak i en sak serien, som ikke inneholder beslektede individer. Hvis saken serien har relateds, da teknisk du ikke har fått en objektiv beregning av P(A|D).,
- Hvis en sykdom er dødelig, så det er vanskelig å få tak i en befolkning kontroll-serien som ikke minst noe utarmet av personer med varianter som forårsaker at sykdom. Så da trenger du ikke en objektiv beregning av P(A) enten.
- Sammenligninger av saken og kontroll allelet frekvens er sårbare for confounding av befolkningen lagdelingen. I prion studien, hadde vi ikke har genome-wide SNP-data om saker, så det var ingen vei til perfekt kontroll for dette.,
- Mange kausale varianter for sjeldne sykdommer som er så sjeldne at selv med ExAC, vi ennå ikke har tilstrekkelig presise estimater av allelet frekvens for å gi bedre enn et grovt svar.
Med alle som sa, population-based allelet frekvens estimering er fortsatt en god måte å få grov ordre-av-magnitude estimater av penetrance og til å utføre fornuft sjekker om en genetisk variant plausibly kan være utløsende for en sjelden sykdom.