RNA-sekvensering (RNA-seq) har et bredt spekter av applikasjoner, og det er ingen optimal rørledning for alle tilfeller. Vi gjennomgå alle de store skritt i RNA-seq-data-analyse, herunder kvalitetskontroll, lese justering, kvantifisering av genet, og transkripsjonen nivåer, differensiert genuttrykk, funksjonelle profilering, og avansert analyse. De vil bli diskutert senere.
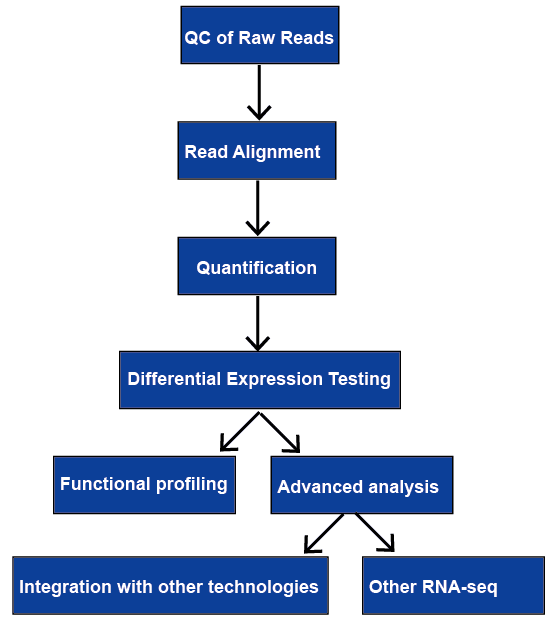
Figur 1. Den generelle arbeidsflyt av RNA-seq analyse.,
kvalitetskontroll av raw leser
kvalitetskontroll av RNA-seq raw leser består av analyse av sekvensen kvalitet, GC-innhold, adapter innhold, overrepresentert k-mers, og duplisert leser, dedikert til å oppdage sekvensering feil, smitte, og PCR-gjenstander. Les kvalitet avtar mot 3′ enden av lyder, baser med lav kvalitet, derfor bør de fjernes for å forbedre mappability., I tillegg til kvaliteten på rådata, kvalitetskontroll av raw leser også omfatter analyse av lese-justering (les ensartethet og GC-innhold), kvantifisering (3′ bias, biotypes, og lav-teller), og reproduserbarhet (korrelasjon, principal component analyse, og batch-effekter).
Tabell 1. Verktøy for kvalitetskontroll av RNA-seq raw-leser.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Uavhengig av om et genom eller transcriptome referanse er tilgjengelig, leser kan kart unikt eller tilordnes til flere stillinger i referansen, som er referert til som multi-kartlagt leser eller multireads. Genomisk multireads er vanligvis på grunn av repeterende sekvenser eller delt domener av paralogous gener. Transcriptome multi-kartlegging oppstår ofte på grunn av genet isoforms. Derfor, transkripsjon identifisering og kvantifisering er viktige utfordringer for alternativt uttrykte gener., Når en referanse ikke er tilgjengelig, RNA-seq leser er samlet de novo bruke pakker som SOAPdenovo-Trans, Oaser, Trans-Avgrunnen, eller Treenighet. PE strand-spesifikke og lange lyder er foretrukket fordi de er mer informative. Nye lang-les-teknologier, for eksempel PacBio SMRT sekvensering og Nanopore sekvensering, kan generere full-lengde transkripsjoner for de fleste gener.
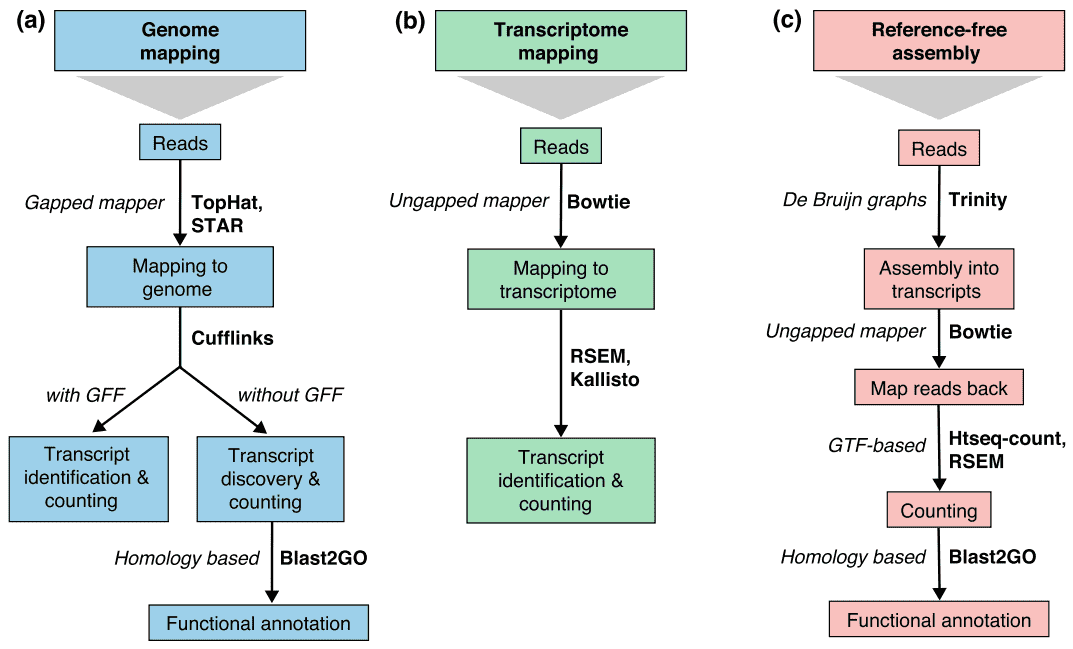
Figur 2. Tre grunnleggende strategier for RNA-seq les kartlegging (Conesa et al. 2016)., Forkortelser: GFF, Generell Funksjon Format; GTF, gene transfer format; RSEM, RNA-seq av Forventning Maksimering.
Tabell 2. Sammenligning av genom-basert og de novo montering strategier for RNA-seq analyse.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Mansjettknapper | Designet for å dra nytte av PE leser, og kan bruke GTF informasjon til å identifisere uttrykt transkripsjoner, eller kan antyde transkripsjoner de novo fra kartlegging data alene. |
| RSEM | Kvantifisere uttrykk fra transcriptome kartlegging. Tildele multi-kartlegging leser blant transkripsjon og utgang innen-eksempel normaliserte verdier som er korrigert for sekvensering skjevheter., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
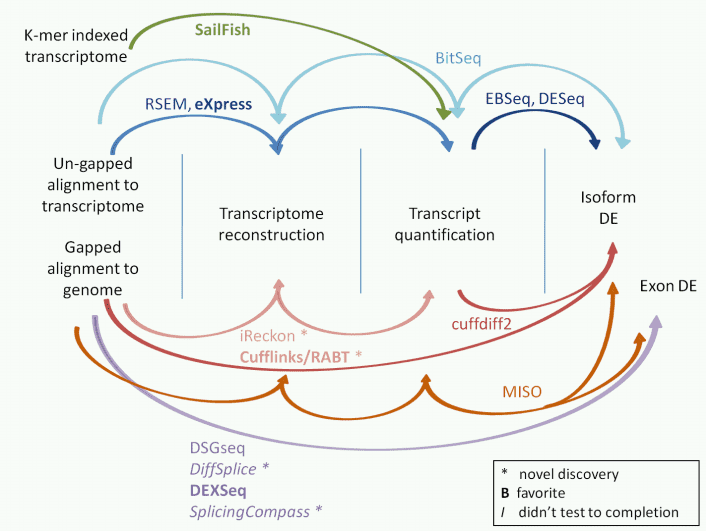
Figure 3. The tools for isoform expression quantification.,
Differensial uttrykk testing
Differensial uttrykk testing brukes til å vurdere om ett gen er differentially uttrykt i en tilstand i forhold til den andre(s). Normalisering metoder må være vedtatt før sammenligne forskjellige prøver. RPKM og TPM normalisere unna den viktigste faktoren, sekvensering dybde. TMM, DESeq, og UpperQuartile kan ignorere svært variabel og/eller sterkt uttrykt funksjoner., Andre faktorer som forstyrrer intra-eksempel sammenligninger innebære transkripsjonen lengde, stedsbestemt skjevheter i dekning, gjennomsnittlig fragment størrelse, og GC-innhold, som kan bli normalisert med verktøy, slik som DESeq, edgeR, baySeq, og NOISeq. Batch effekter kan fortsatt være tilstede etter normalisering, som kan reduseres ved riktig eksperimentell design, eller fjernes ved hjelp av metoder som KAMP eller ARSyN.
Tabell 5. Normalisering verktøy for differensial uttrykk testing.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Flere bioinformatikk-verktøy som er utviklet for å oppdage SOM fra eksperimentelle data. Sammenligning av disse to verktøyene ved hjelp av RNA-seq data ble utført av Ding i 2017, og resultatene er vist i Tabell 7. De har vist at TopHat og dens nedstrøms verktøyet, FineSplice, er den raskeste verktøy, mens PASTAEN er den tregeste programmet. Videre, AltEventFinder kan oppdage det høyeste antallet av veikryss, og RSR oppdager det laveste antall knutepunkter. Andre verktøy, for eksempel TopHat, er sannsynlig å oppdage falske positive meldinger., Av de to verktøy som gjenkjenner differentially skjøtes isoforms, rMATS er raskere enn rSeqDiff men oppdager mindre differentially skjøtes isoforms enn rSeqDiff.
Tabell 7. Oppdaget SOM typer eller differentially skjøtes isoforms av disse verktøyene (Ding et al. 2017).,
Visualisering
Det er mange bioinformatikk-verktøy for visualisering av RNA-seq data, inkludert genom nettlesere, slik som ReadXplorer, UCSC nettleser, Integrerende Genomics Viewer (IGV), Genom Kart, Savant, verktøy spesielt designet for RNA-seq data, for eksempel RNAseqViewer, samt noen pakker for differensiert genuttrykk analyse som gjør det mulig å visualisere, for eksempel DESeq2 og DEXseq i Bioconductor. Pakker, slik som CummeRbund og Sashimi tomter, har også blitt utviklet for visualisering-eksklusiv formål.,
Funksjonell Profilering
siste skritt i en standard transcriptomics studien er generelt karakterisering av molekylære funksjoner eller stier som differentially uttrykte gener er involvert. Gene Ontology, Bioconductor, DAVID, eller Babelomics inneholde merknaden data for de fleste modell arter, som kan brukes for funksjonell bruk. Som for romanen transkripsjoner, protein-kodende transkripsjoner kan være funksjonelt forklart ved hjelp av orthology med hjelp av databaser som SwissProt, Pfam, og InterPro., Gene Ontology (GÅ) gjør det mulig for noen exchangeability av funksjonelle informasjon på tvers av orthologs. Blast2GO er et populært verktøy som tillater massiv markering av fullført transcriptome mot en rekke databaser og kontrollert vocabularies. Den Rfam database inneholder mest godt preget RNA familier som kan brukes for funksjonell markering av lange ikke-kodende RNAs.
Avansert analyse
avansert analyse av RNA-seq omfatter vanligvis andre RNA-seq og integrasjon med andre teknologier, som er skissert i Figur 4., Mer informasjon på anvendelser av RNA-seq, kan du se denne artikkelen Programmer av RNA-Seq.
Figur 3. Avansert analyse av RNA-seq data.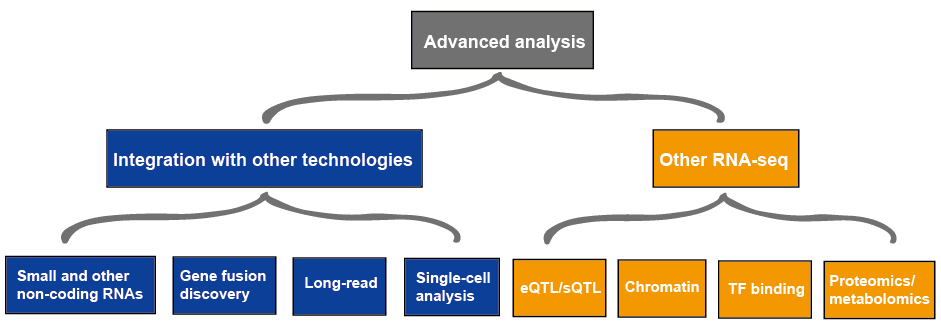
Våre erfarne bioinformatikk forskere er dyktige i å utnytte avansert bioinformatikk-verktøy for å håndtere de mange sekvenser generert av den neste og tredje generasjons sekvensering. Vi leverer både sekvensering og bioinformatikk tjenester for genomics, transcriptomics, epigenomics, mikrobiell genomics, encellede sekvensering, og PacBio SMRT sekvensering.