RNA-sekvensointi (RNA-seq) on laaja valikoima sovelluksia, ja ei ole optimaalinen putki kaikissa tapauksissa. Käymme läpi kaikki tärkeimmät vaiheet RNA-seq-tietojen analysoinnissa, mukaan lukien laadunvalvonta, lukulinjaus, geeni-ja transkriptiotasojen kvantifiointi, differentiaaligeenin ilmentyminen, funktionaalinen profilointi ja kehittynyt analyysi. Niistä keskustellaan myöhemmin.
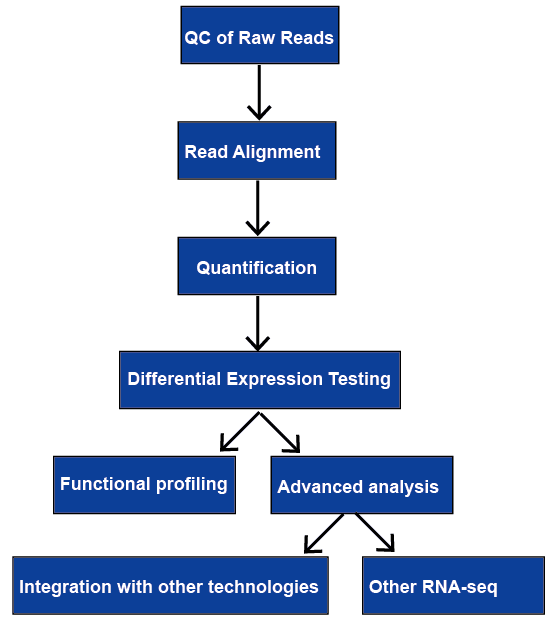
Kuva 1. RNA-seq-analyysin yleinen työnkulku.,
raw readsin laadunvalvonta
RNA-seq raw readsin laadunvalvonta koostuu sekvenssilaadun, GC-sisällön, Muuntimen sisällön, yliedustetun k-Mersin ja monistetun Readingin analysoinnista, joka on omistettu sekvenssivirheiden, kontaminaatioiden ja PCR-artefaktien havaitsemiseen. Lukulaatu laskee kohti 3 ’ loppuun lukee, emäkset huonolaatuisia, siksi ne olisi poistettava parantaa mappability., Lisäksi laadukkaita raaka-tietoja, laadunvalvonta raaka-lukee myös analyysi lukea linjaus (lue yhdenmukaisuus ja GC-pitoisuus), kvantifiointi (3’ bias, biotyyppejä, ja alhainen määrä), ja uusittavuus (korrelaatio, principal component analysis, ja erän vaikutukset).
Taulukko 1. Työkalut laadunvalvontaan RNA-seq raw lukee.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Riippumatta siitä, onko genomi-tai transcriptome-viittaus saatavilla, reades voi kartoittaa yksilöllisesti tai olla osoitettavissa useampaan kohtaan viittauksessa, joista käytetään nimitystä multi-mapped reads tai multireads. Genomiset multireadit johtuvat yleensä paralogisten geenien toistuvista sekvensseistä tai jaetuista domeeneista. Transcriptome multi-kartoitus syntyy useammin geenien isoformien takia. Siksi transkription tunnistaminen ja kvantifiointi ovat tärkeitä haasteita vaihtoehtoisesti ilmaistuille geeneille., Kun viittaus on ole saatavilla, RNA-seq lukee kootaan de novo käyttäen paketteja, kuten SOAPdenovo-Trans, Keitaita, Trans-Kuiluun, tai Trinity. PE strand-spesifiset ja pitkät lukemat ovat suositeltavia, koska ne ovat informatiivisempia. Kehittyvien pitkä-lue teknologioita, kuten PacBio SMRT sekvensointi ja Nano-sekvensointi, voi tuottaa kokoillan selostukset useimmat geenit.
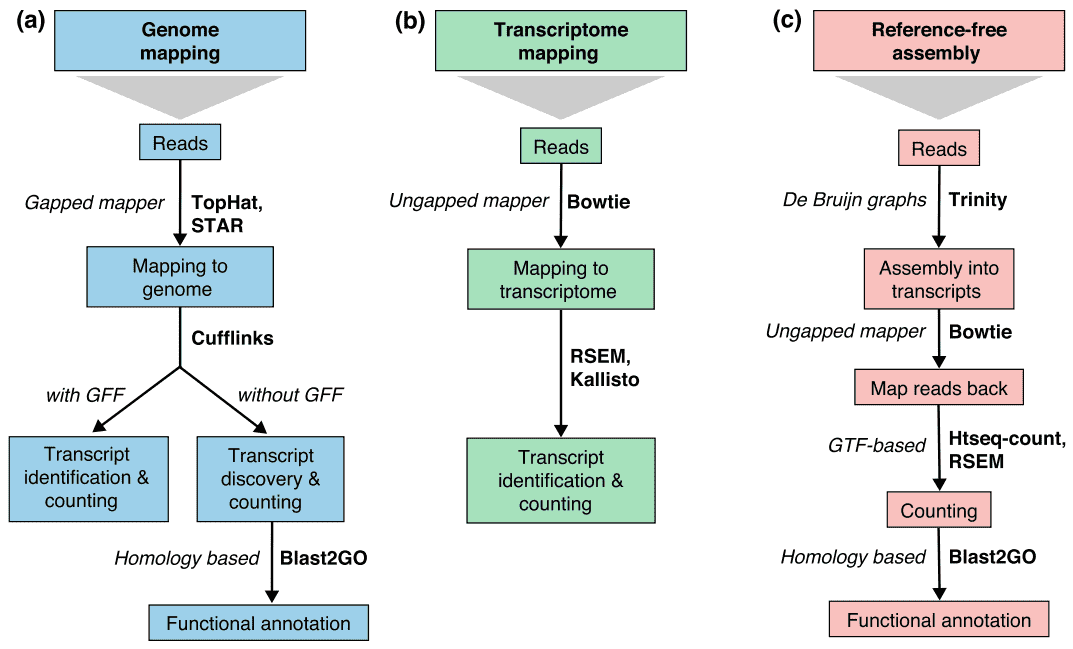
Kuva 2. Kolme perusstrategiaa RNA-seq lukea kartoitus(Conesa et al. 2016)., Lyhenteet: GFF, yleinen Ominaisuusmuoto; GTF, geeninsiirtomuoto; RSEM, RNA-seq odotuksen maksimoinnin avulla.
Taulukko 2. Genomipohjaisten ja de novo assembly-strategioiden vertailu RNA-seq-analyysiin.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| kalvosinnapit | on suunniteltu hyödyntämään PE readersia, ja se voi käyttää GTF-tietoja ilmaistujen transkriptioiden tunnistamiseen, tai se voi päätellä de Novon pelkästään kartoitustietojen perusteella. |
| RSEM | Määrittää ilmaisun transcriptome kartoitus. jakaa monikartoitus lukee transkriptiossa ja ulostulo otoksen sisällä normalisoidut arvot korjattu jaksottamista varten., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
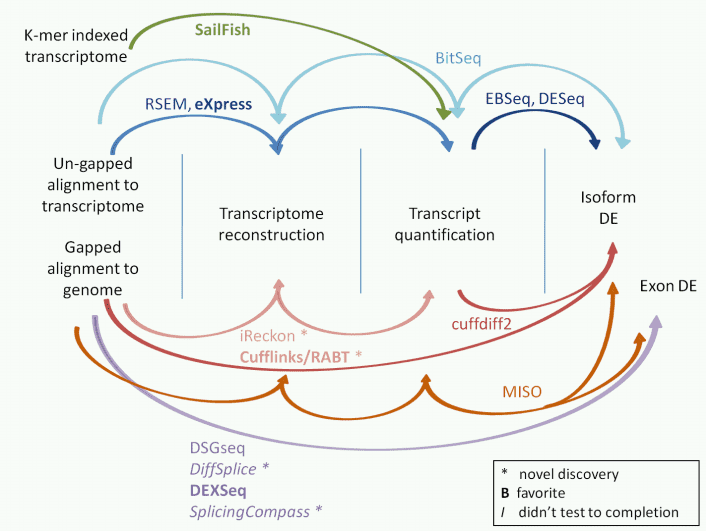
Figure 3. The tools for isoform expression quantification.,
Ero ilmaisun testaus
Ero ilmaisun testaus on tapana arvioida, jos yksi geeni on eri tavoin ilmaistu yhdellä ehdolla verrattuna muihin(s). NormalisointiMenetelmät on otettava käyttöön ennen eri näytteiden vertailua. RPKM ja TPM normalisoivat pois tärkeimmän tekijän, sekvensointisyvyyden. TMM, DESeq ja Upperkartile voivat sivuuttaa erittäin vaihtelevia ja / tai erittäin ilmaistuja ominaisuuksia., Muita tekijöitä, jotka häiritsevät intra-näyte vertailujen mukaan transkriptio pituus, asentohuimaus harhat kattavuus, keskimääräinen fragmentti koko, ja GC sisältöä, joka voi olla normalisoitu työkaluja, kuten DESeq, särmäysautomaatti, baySeq, ja NOISeq. Erän vaikutukset voivat olla vielä läsnä normalisoinnin jälkeen, mikä voidaan minimoida asianmukaisella kokeellisella suunnittelulla tai poistaa menetelmillä, kuten COMBAT tai ARSyN.
Taulukko 5. Normalisointi työkalut differentiaali lauseke testaus.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Kokeellisen tiedon havaitsemiseksi on kehitetty useita bioinformatiikan työkaluja. Näiden tunnistusvälineiden vertailu RNA-seq-tietojen avulla toteutettiin Ding-menetelmällä vuonna 2017, ja tulokset on esitetty taulukossa 7. He ovat osoittaneet, että TopHat ja sen jatkojalostustyökalu FineSplice ovat nopeimpia työkaluja, kun taas PASTA on hitain ohjelma. Lisäksi AltEventFinder pystyy havaitsemaan eniten liittymiä, ja RSR havaitsee vähiten liittymiä. Muut työkalut, kuten TopHat, todennäköisesti havaitsevat vääriä positiivisia., Rmats on nopeampi kuin rSeqDiff, mutta se havaitsee vähemmän eriytyneitä isoformeja kuin rSeqDiff.
Taulukko 7. Havaittu tyyppeinä tai differentiaalisesti yhdistettyinä näiden työkalujen isoformeina (Ding et al. 2017).,
Visualisointi
On olemassa monia bioinformatiikan työkaluja visualisointi RNA-seq tietoja, kuten genomin selaimet, kuten ReadXplorer, UCSC selain, Integroiva Genomics Viewer (IGV), Genomin Karttoja, Savant, työkaluja suunniteltu erityisesti RNA-seq tietoja, kuten RNAseqViewer, sekä joitakin paketteja ero geenien ilmentyminen analyysi, joka mahdollistaa visualisoinnin, kuten DESeq2 ja DEXseq vuonna Bioconductor. Paketteja, kuten CummeRbund-ja Sashimi-tontteja, on kehitetty myös visualisointiin-eksklusiivisiin tarkoituksiin.,
Toiminnallinen Profilointi
viimeisin askel standardin transcriptomics tutkimus on yleensä luonnehdinta molekyyli toimintoja tai tapoja, jotka eri tavoin ilmaistuna geenit ovat mukana. Gene Ontologia, Bioconductor, DAVID, tai Babelomics sisältävät merkintä tiedot useimmille malli lajeja, joita voidaan käyttää toiminnallinen kommentointi. Uusien transkriptioiden osalta proteiinikoodausranskriptioita voidaan toiminnallisesti merkitä ortologian avulla tietokantojen kuten SwissProt, Pfam ja InterPro avulla., Gene Ontology (GO) mahdollistaa funktionaalisen tiedon jonkin verran vaihdettavuutta ortologien välillä. Blast2GO on suosittu työkalu, joka mahdollistaa massiivisen huomautuksen täydellinen transkriptome vastaan erilaisia tietokantoja ja valvottuja sanastoja. Rfam-tietokanta sisältää parhaiten tunnettuja RNA-perheitä, joita voidaan käyttää pitkän koodaamattoman RNAs: n toiminnalliseen selitykseen.
Lisäasetukset-analyysi
lisäasetukset-analyysi RNA-seq sisältää yleensä muita RNA-seq ja integrointi muihin teknologioihin, jotka on esitetty Kuvassa 4., Lisätietoja RNA-seq: n sovelluksista, katso tämä artikkeli RNA-Seq: n Sovellukset.
kuva 3. RNA-seq-tietojen edistynyt analyysi.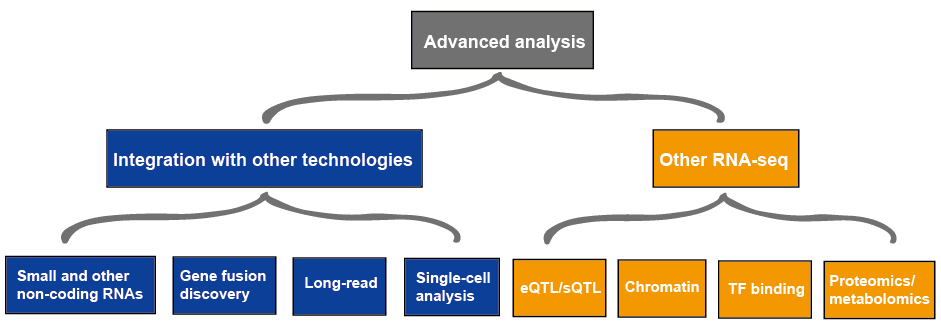
kokenut bioinformatiikan tutkijat ovat taitavia hyödyntämällä kehittyneitä bioinformatiikan työkaluja käsitellä lukuisia sarjoja syntyy seuraavan ja kolmannen sukupolven sekvensointi. Tarjoamme sekä sekvensointi ja bioinformatiikka palvelut genomiikka, transcriptomics, epigenomics, mikrobien genomiikka, yksisoluiset sekvensointi, ja PacBio SMRT sekvensointi.