RNA-Sequenzierung (RNA-seq) hat eine breite Palette von Anwendungen, und es gibt keine optimale Pipeline für alle Fälle. Wir überprüfen alle wichtigen Schritte in der RNA-seq-Datenanalyse, einschließlich Qualitätskontrolle, Leseausrichtung, Quantifizierung von Gen-und Transkriptniveaus, differentieller Genexpression, Funktionsprofilerstellung und erweiterter Analyse. Sie werden später besprochen.
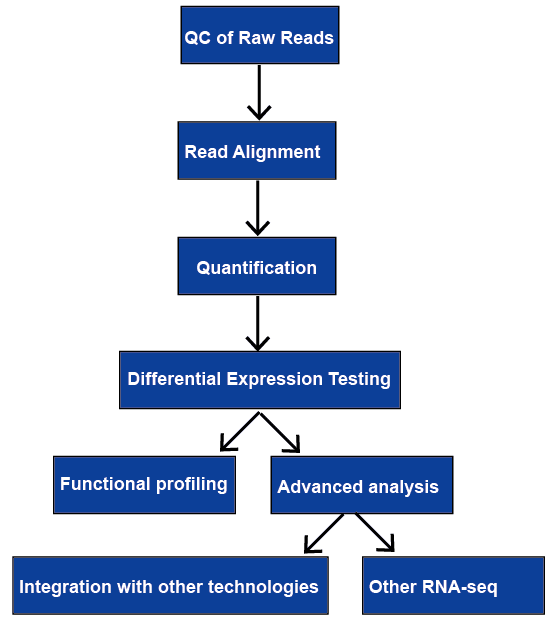
Abbildung 1. Der allgemeine Workflow der RNA-seq-Analyse.,
Qualitätskontrolle von Raw-Lesevorgängen
Die Qualitätskontrolle von RNA-seq-Raw-Lesevorgängen besteht aus der Analyse der Sequenzqualität, des GC-Inhalts, des Adapterinhalts, der überrepräsentierten k-mers und der duplizierten Lesevorgänge, die der Erkennung von Sequenzierungsfehlern, Kontaminationen und PCR-Artefakten gewidmet sind. Die Lesequalität nimmt gegen Ende der Lesevorgänge von 3 ‚ ab, Basen mit geringer Qualität sollten daher entfernt werden, um die Zuordenbarkeit zu verbessern., Neben der Qualität der Rohdaten umfasst die Qualitätskontrolle von Rohdaten auch die Analyse der Leseausrichtung (Leseinformität und GC-Gehalt), Quantifizierung (3‘ Bias, Biotypen und niedrige Zählungen) und Reproduzierbarkeit (Korrelation, Hauptkomponentenanalyse und Chargeneffekte).
Tabelle 1. Die Werkzeuge zur Qualitätskontrolle von RNA-seq-Rohdaten.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Unabhängig davon, ob eine Genom-oder Transkriptomreferenz verfügbar ist, können Lesevorgänge eindeutig zugeordnet oder mehreren Positionen in der Referenz zugewiesen werden, die als Multi-Mapped-Lesevorgänge oder Multireads bezeichnet werden. Genomische Multireaden sind im Allgemeinen auf sich wiederholende Sequenzen oder gemeinsame Domänen paraloger Gene zurückzuführen. Transkriptom-Multi-Mapping entsteht häufiger aufgrund von Gen-Isoformen. Daher sind die Identifizierung und Quantifizierung von Transkripten wichtige Herausforderungen für alternativ exprimierte Gene., Wenn eine Referenz nicht verfügbar ist, werden RNA-seq-Lesevorgänge mithilfe von Paketen wie SOAPdenovo-Trans, Oasen, Trans-ABySS oder Trinity de novo zusammengestellt. PE-strangspezifische und lange Lesevorgänge werden bevorzugt, da sie informativer sind. Aufkommende Long-Read-Technologien wie PacBio SMRT-Sequenzierung und Nanopore-Sequenzierung können Transkripte in voller Länge für die meisten Gene erzeugen.
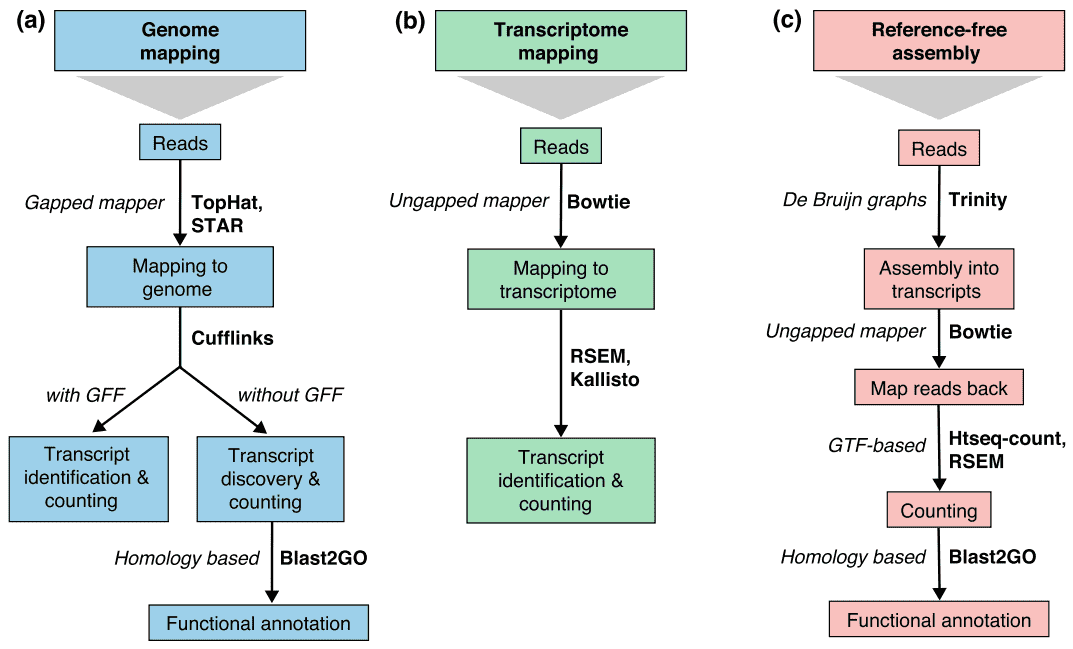
Abbildung 2. Drei grundlegende Strategien für die RNA-seq-read-mapping (Conesa et al. 2016)., Abkürzungen: GFF, Allgemeines Merkmalsformat; GTF, Gentransferformat; RSEM, RNA-seq durch Erwartungsmaximierung.
Tabelle 2. Der Vergleich der Genom-basierten und de-novo-Montage Strategien für die RNA-seq-Analyse.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Entwickelt, um die vorteile von PE liest, und kann GTF informationen zu identifizieren ausgedrückt transkripte, oder können ableiten transkripte de novo von die mapping daten allein. | |
| RSEM | Quantifizierung der expression von Transkriptom-mapping. Zuweisen Multi-Mapping liest unter Transkript und Ausgabe innerhalb der Probe normalisierte Werte für Sequenzierungsverzerrungen korrigiert., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
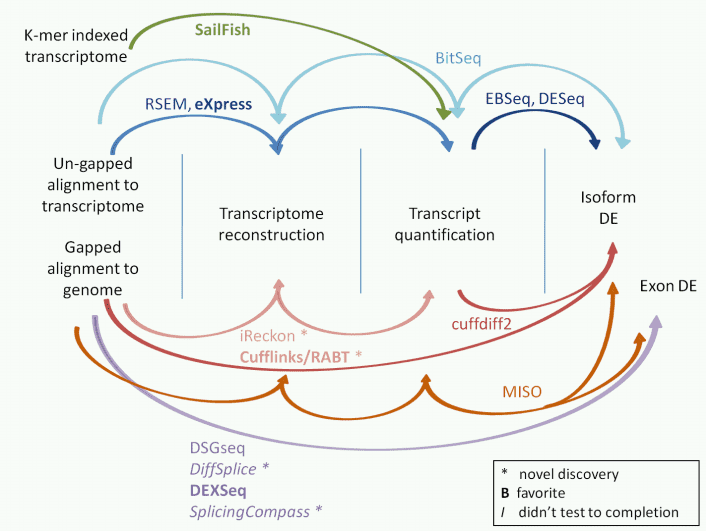
Figure 3. The tools for isoform expression quantification.,
Differentielle expression testing
Differentielle expression Prüfung wird verwendet, um festzustellen, ob ein gen ist differentiell exprimiert in einem Zustand im Vergleich zu den anderen(s). Normalisierungsmethoden müssen angewendet werden, bevor verschiedene Proben verglichen werden. RPKM und TPM normalisieren weg der wichtigste faktor, sequenzierung tiefe. TMM, DESeq und Oberquartil können hochvariable und/oder hoch exprimierte Funktionen ignorieren., Andere Faktoren, die den Vergleich innerhalb der Stichprobe beeinträchtigen, umfassen die Länge der Transkripte, Positionsverzerrungen in der Abdeckung, die durchschnittliche Fragmentgröße und den GC-Inhalt, die mit Tools wie DESeq, edgeR, baySeq und NOISeq normalisiert werden können. Batch-Effekte können nach der Normalisierung noch vorhanden sein, die durch geeignetes experimentelles Design minimiert oder durch Methoden wie COMBAT oder ARSyN entfernt werden können.
Tabelle 5. Die Normalisierung Werkzeuge für die differentielle expression testen.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Mehrere Bioinformatik-Tools wurden entwickelt, um AS aus experimentellen Daten zu erkennen. Der Vergleich dieser Nachweiswerkzeuge mit RNA-seq-Daten wurde 2017 von Ding durchgeführt und die Ergebnisse sind in Tabelle 7 dargestellt. Sie haben gezeigt, dass TopHat und sein nachgelagertes Tool FineSplice die schnellsten Werkzeuge sind, während PASTA das langsamste Programm ist. Darüber hinaus kann AltEventFinder die höchste Anzahl von Kreuzungen erkennen, und RSR erkennt die niedrigste Anzahl von Kreuzungen. Andere Tools wie TopHat erkennen wahrscheinlich falsch positive., Von den beiden Tools, die unterschiedlich gespleißte Isoformen erkennen, ist rMATS schneller als rSeqDiff, erkennt jedoch weniger unterschiedlich gespleißte Isoformen als rSeqDiff.
Tabelle 7. Erkannt als Typen oder differentiell gespleißte Isoformen dieser Werkzeuge (Ding et al. 2017).,
Visualisierung
Es gibt viele Bioinformatik-Tools für die Visualisierung von RNA-seq-Daten, einschließlich Genom-Browsern wie ReadXplorer, UCSC Browser, Integrative Genomics Viewer (IGV), Genom Maps, Savant, speziell für RNA-seq-Daten entwickelte Tools wie RNAseqViewer sowie einige Pakete für die differentielle Genexpressionsanalyse, die die Visualisierung ermöglichen, wie DESeq2 und DEXseq in Bioconductor. Pakete, wie CummeRbund und Sashimi Plots, wurden ebenfalls für Visualisierungs-exklusive Zwecke entwickelt.,
Functional Profiling
Der letzte Schritt in einer Standard-Transkriptomik-Studie ist im Allgemeinen die Charakterisierung der molekularen Funktionen oder Wege, an denen differentiell exprimierte Gene beteiligt sind. Gene Ontology, Bioconductor, DAVID oder Babelomics enthalten Annotationsdaten für die meisten Modellarten, die zur funktionellen Annotation verwendet werden können. Bei neuartigen Transkripten können proteincodierende Transkripte mithilfe von Datenbanken wie SwissProt, Pfam und InterPro funktional orthologisch kommentiert werden., Die Genontologie (GO) ermöglicht einen gewissen Austausch funktioneller Informationen über Orthologen hinweg. Blast2GO ist ein beliebtes Tool, das eine massive Annotation des vollständigen Transkriptoms gegen eine Vielzahl von Datenbanken und kontrollierten Vokabularen ermöglicht. Die Rfam-Datenbank enthält die am besten charakterisierten RNA-Familien, die für die funktionelle Annotation langer nicht kodierender RNAs verwendet werden können.
Erweiterte Analyse
Die erweiterte Analyse von RNA-seq umfasst in der Regel andere RNA-seq und Integration mit anderen Technologien, die in Abbildung 4., Weitere Informationen zu Anwendungen von RNA-seq finden Sie in diesem Artikel Anwendungen von RNA-Seq.
Abbildung 3. Die erweiterte Analyse von RNA-seq Daten.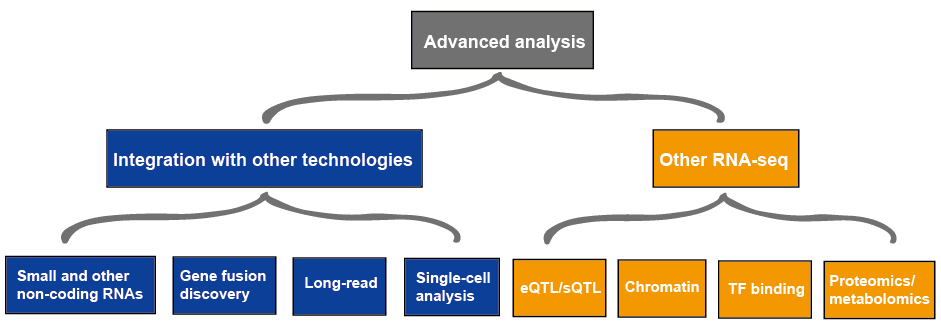
Unsere erfahrenen Bioinformatiker sind in der Verwendung der fortschrittlichen Bioinformatik-Tools, um mit den zahlreichen Sequenzen, die durch die nächste und dritte Generation Sequenzierung. Wir bieten sowohl Sequenzierungs-als auch Bioinformatikdienste für Genomik, Transkriptomik, Epigenomik, mikrobielle Genomik, Einzelzellsequenzierung und PacBio SMRT-Sequenzierung an.