Förra månaden var jag inbjuden att tala vid centrum för Mendels Genomik (CMG) Analys och metodutveckling möte om ”populationsbaserad uppskattning av penetrans i sällsynta sjukdom”. Här är blogginlägget version av my talk.
vad är penetrance och varför bryr vi oss?
Penetrance är sannolikheten att utveckla en viss sjukdom med tanke på en viss genotyp., Man kan tala om åldersberoende penetrance, så andelen personer med genotypen som utvecklar sjukdomen vid 40 års ålder, vid 50 års ålder och så vidare; Jag talar vanligtvis när det gäller livstidsrisk, vilket betyder sannolikheten att du någonsin utvecklar sjukdomen innan du dör. Inneboende i detta är att för vuxna sjukdomar kan livstidsrisken aldrig vara 100%, för att du alltid kan dö av något annat först.
Penetrance är enormt viktigt för individer som genomgår prediktiv genetisk testning-många människors första fråga är, ” betyder det att jag definitivt får sjukdomen?”., Ändå är det ofta mycket svårt att komma med en fast uppskattning av penetrance.
traditionella metoder för att uppskatta penetrance
i en idealisk värld skulle det rätta sättet att uppskatta penetrance vara att från födseln fastställa en stor kohort av personer med en viss genotyp, följa dem tills alla har dött av något eller annat, och fråga sedan hur många som någonsin utvecklat sjukdomen innan de dog. Eftersom genotypningsteknik uppfanns för mindre än en människas livstid sedan, har detta aldrig gjorts för någon sjukdom.
istället använder forskare ofta familjebaserade metoder för att uppskatta penetrance., En typisk studie skulle titta på alla som har observerats med den givna genotypen, och fråga hur många har sjukdomen, eller hur många har sjukdomen vid en viss ålder. Familjebaserade metoder lider av genomgripande bestämningsfördomar ., fastställd på grundval av att presentera med sjukdom
som ett exempel på denna sista punkt, i genetisk prionsjukdom, väljer endast 23% av riskpersoner prediktiv genetisk testning och i stamtavla data som jag har hade tillgång till , visste vi genotyper av endast 22% av risk individer .,
alla faktorer som anges ovan fungerar i samma riktning och tenderar att blåsa upp sin uppskattning av penetrance.
forskare har länge varit medvetna om dessa problem och har föreslagit några lösningar. Som ett exempel innebär kin-kohort-metoden att fastställa friska individer slumpmässigt från en population, genotyping dem, ta en familjehistoria och jämföra överlevnadskurvor hos sina första graders släktingar., Detta är en mycket smart lösning, men det bygger på att kunna fastställa ett tillräckligt stort antal personer med en sjukdomsframkallande genotyp utan att fastställa förekomsten av sjukdom. Så det fungerade för BRCA1 och BRCA2 varianter i amerikanska Ashkenazi judar, men för många ovanligare genetiska förhållanden, det är opraktiskt, eftersom du skulle behöva rekrytera tiotals eller hundratusentals människor att hitta även en individ med en genotyp av intresse.,
befolkningsbaserade metoder
av alla skäl som beskrivs ovan är det mycket användbart att ha ortogonala, befolkningsbaserade metoder för att ställa frågor om penetrance. Den första viktiga insikten här är att en helt penetrerande genetisk variant inte ska vara vanligare i befolkningen än den sjukdom som den orsakar. Att tillämpa denna logik i praktiken innebär att du behöver bra uppskattningar av allelfrekvens även för ovanliga varianter, och det har varit svårt att komma fram tills nyligen. ExAC, en databas med genetisk variation i 60,706 mänskliga exomer, erbjuder nya möjligheter ., Många individer i ExAC fastställdes som fall eller kontroller för olika vanliga, komplexa sjukdomar, men ingen fastställdes för Mendelisk sjukdom, så ExAC är en bra referensdatabas för att studera de flesta genetiska sjukdomar.
genom att tillhandahålla allelfrekvensinformation i den allmänna befolkningen har ExAC, liksom tidigare referensdatabaser som ESP, gjort det klart att klinisk genetik har ett stort problem: många varianter rapporterade att orsaka genetisk sjukdom faktiskt inte orsakar genetisk sjukdom, eller åtminstone inte för det mesta.,
två databaser — Hgmd och ClinVar — samlar påståenden från litteraturen och från kliniska laboratorier som anger att en viss genetisk variant orsakar en viss genetisk sjukdom. Till sist fanns det över 100 000 unika reportedy sjukdomsframkallande genetiska varianter i dessa databaser. Den genomsnittliga personen i ExAC har 54 av dem . Självklart har den genomsnittliga personen faktiskt inte 54 genetiska sjukdomar., Naturligtvis är mycket av detta överskott orsakas av ett litet antal vilt högfrekventa varianter som uppenbarligen inte orsakar någon genetisk sjukdom, och mycket av det kan vara enligt uppgift recessiva varianter som finns i ett heterozygot tillstånd i ExAC. Men även om vi bara tittar på varianter i dominerande sjukdomsgener vid en allelfrekvens på<1% ser vi fortfarande 0,89 enligt uppgift patogena varianter per person, och det är uppenbarligen inte fallet att ~90% av befolkningen har en dominerande genetisk sjukdom., Så över allelfrekvensspektrumet finns det många enligt uppgift patogena varianter som inte är så patogena. När Anne O ’ Donnell och jag tittade på de enligt uppgift patogena varianterna med de högsta allelfrekvenserna i ExAC, och frågade hur de hade lyckats felklassificeras som patogena, fann vi att för det mesta problemet spåras till ett papper i litteraturen som hade gjort ett påstående om patogenicitet baserat på otillräckliga bevis.
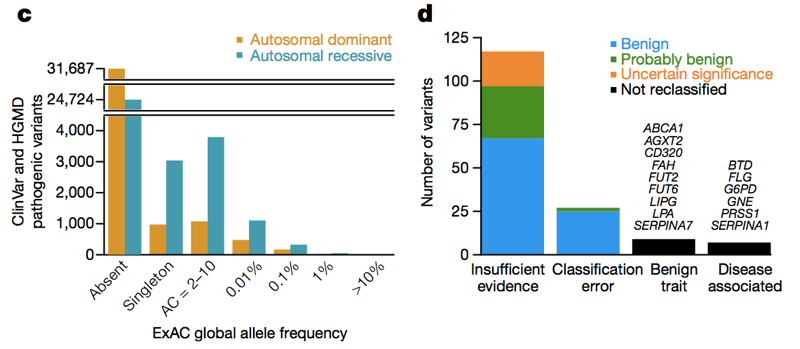
ovan: figurerna 3c och 3D från ., Över allelfrekvensspektrumet och i både dominerande och recessiva sjukdomsgener finns det många enligt uppgift patogena varianter som förekommer i ExAC. Av hög (>1%) frekvens enligt uppgift patogena varianter, några är genuint patogena, vissa är genuint drag-associerade men egenskapen är godartad, och vissa är fel i anteckning i databaser — men majoriteten är baserade på litteratur med otillräckliga bevis.,
allelfrekvensinformation från ExAC har nu gjort det möjligt att omklassificera över 200 genetiska varianter från patogena till godartade, troligen godartade eller av osäker betydelse . Dessa typer av omklassificeringar utlöser ibland pushback från de ursprungliga författarna som föreslog att en variant orsakar en genetisk sjukdom, som kan hävda att en variant fortfarande kan vara patogen, men med ofullständig penetrans. Men hur ”ofullständig” kan ofullständig penetrans vara?, Vi måste få kvantitativa, för om livstidsrisken är högst 1%, är det fortfarande rimligt att säga att en variant ”orsakar” en genetisk sjukdom eller är ”patogen”? Medan allelfrekvensinformation aldrig kan bevisa att en variant inte har någon koppling till sjukdom, kan den sätta gränser för vad den möjliga penetransen kan vara, och i många fall, även för ganska sällsynta varianter, är det möjligt att visa att det inte finns något sätt att en variant ger en risknivå någonstans nära 100%.,
för att få kvantitativa måste vi utöka vår tidigare observation-att en helt penetrerande genetisk variant inte borde vara vanligare i befolkningen än den sjukdom som den orsakar. Detta är alla enkla matematik och population genetik, men det är alltför ofta inte tillämpas i praktiken. Här är två sätt vi kan tänka på allelfrekvens när man gör slutsatser om patogenicitet och penetrance.
maximal trovärdig allelfrekvens
säg att du studerar exomen hos en patient med mendelisk sjukdom och försöker identifiera orsaksvarianten., Min kollega James Ware har utarbetat en strategi för filtrering som exome mot allele frekvensinformation i ExAC, dra nytta av följande logik., Den maximala allelfrekvensen som är rimlig för en variant för att orsaka en dominerande genetisk sjukdom är lika med förekomsten av sjukdomstiderna allel heterogeniteten (andel fall som kan hänföras till en variant) dividerad med penetrance (mindre penetrerande varianter kan vara vanligare), dividerad med 2 (eftersom vi är diploid):
\
till exempel orsakar prionsjukdom i 1 i 5000 dödsfall, och den vanligaste varianten (E200K) finns i 5% av fallen, så en 100% penetrerande variant kan inte möjligen ha allelfrekvens större än 0 , 0005% (1 av 200 000) ., Kardiomyopati påverkar 1 av 500 personer, den vanligaste varianten finns i <2% av fallen, så en 50% penetrerande variant kan inte ha en allelfrekvens som är större än 0,004% . Formeln för recessiva sjukdomar är ett hack mer komplicerat men James har också utarbetat det och det beskrivs i .
så medan historiskt har människor ofta filtrerat ut varianter med en allelfrekvens >0.1% när man försöker identifiera orsaken till en dominerande sjukdom , kan vi faktiskt vara mycket strängare., Förbehållet är att vid låga allelantal är vår förmåga att uppskatta allelfrekvensen begränsad av provtagningsvarians. Om vi till exempel tittar på varianter som ses med en 1-procentig allelfrekvens bland européerna i ESP, har dessa varianter också en 1-procentig frekvens bland ExAC-européer. Men varianter med en 0,1% frekvens i ESP tenderar att vara lite sällsynta i ExAC, och de flesta singletons (varianter som ses exakt en gång i ESP) återkommer inte en andra gång i ExAC.
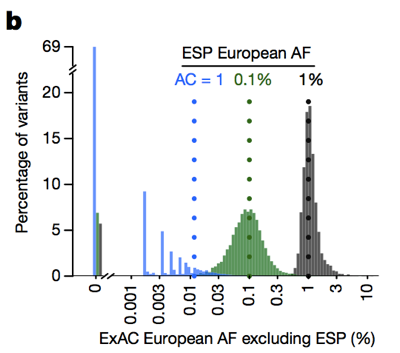
ovan: figur 3b från . Ju lägre allelantalet är, desto mindre bra en uppskattning av allelfrekvensen ger den.,
ju lägre allelantalet desto mer konservativt måste vi vara. Vi har utarbetat ett ramverk för att göra detta med den 95% övre gränsen för Poisson-fördelningen på hur många alleler som kan observeras vid en viss frekvens och har förutbestämda värden för alla ExAC (tillgängliga på FTP) som du kan använda-Läs mer om metoderna i . James har också skapat denna praktiska webbapp som låter dig utforska vad ”maximal trovärdig allelfrekvens” borde vara för din sjukdom av intresse.,
inneboende i detta tillvägagångssätt är att ju lägre penetrationen av en variant, desto högre frekvens kan det ha i den allmänna befolkningen. Men du måste också räkna med att om penetrance är ganska låg, säg mindre än 10%, är den kliniska nyttan av den varianten också låg. James och Nicky Whiffin har presenterat data som visar att nästan alla av den kliniska nyttan av sekvensering i kardiomyopati kommer från varianter med en frekvens på <0.001% — mer vanliga varianter sammantaget bidrar mycket lite, om något, är risken .,
uppskattning och gränser för livstidsrisk
Kom ihåg att penetrance är sannolikheten för sjukdom som ges en viss genotyp. Eller, om vi betraktar en allelisk snarare än genotypisk modell, sannolikheten för sjukdom ges en viss allel. Vi kan skriva detta som P(D|A). När vi gör det blir det klart att Bayes ’ sats,
\
var och en av dessa termer har en särskild betydelse:
Observera här att ”befolkningskontroller” betyder en grupp som inte valts ut för närvaron eller frånvaron av sjukdomen. Bara en del av befolkningen.
Så:
\
denna logik är inget nytt., Användningen av Bayes teorem för att uppskatta sjukdomsrisken går åtminstone tillbaka till uppskattningen av risken för cancer hos rökare , och dess tillämpning på genetiken har beaktats nästan lika länge . Men för att denna ekvation ska fungera för sällsynta sjukdomar behöver du ganska bra uppskattningar av fall och befolkningskontroll allelfrekvens, och de har varit svåra att komma fram till tills nyligen. Så tack vare ExAC finns det ett ökande antal situationer där denna ekvation är relevant.
Här är R-koden jag har skrivit (ursprungligen här) för att uppskatta penetrance baserat på denna formel.,
Om du inte vill köra r-koden själv har James Ware implementerat den på fliken ”penetrance” i den här webbappen så att du bara kan ansluta dina nummer till din webbläsare.
för att uppskatta 95% konfidensintervall på penetrance har jag antagit tillvägagångssättet för . Du matar in allelantalet (AC) och antalet individer (N) för fall och kontroller, och den övre gränsen för 95% CI beräknas baserat på den övre 95% CI av binomialfördelningen för fall allelfrekvens och den nedre 95% CI för kontroller., Omvänt är den nedre gränsen för penetrance baserad på den nedre gränsen för fall allelfrekvens och den övre gränsen för kontroll allelfrekvens. Du kan med rätta quibble att eftersom denna formel använder 95% CIs på båda allelfrekvensvärdena är de resulterande konfidensintervallen större än de borde vara. Du kan också med rätta quibble att binomialfördelningen inte är en bra estimator vid låga allelantal, på grund av den bias som illustreras i figur 3B som visas ovan (och jag skulle verkligen aldrig tillämpa denna formel på singletons — varianter observerade endast en gång i ExAC)., Men i slutet av dagen, av skäl som jag ska diskutera närmare slutet av detta inlägg, är denna formel verkligen bäst används för att få en ballpark, storleksordning uppskattning av penetrance. Om du letar efter en extremt exakt punktskattning av penetrance, kommer hela detta tillvägagångssätt förmodligen inte att fungera för dig ändå.
om du omordnar ekvationen är ett annat sätt att tänka på det:
\
det betyder att den ökade risken bland personer med genotyp är proportionell mot förhållandet mellan fall till befolkningskontroll allelfrekvens., Så en variant som ökar risken med 200 gånger bör vara 200 gånger vanligare bland Fallen än i den allmänna befolkningen. (Observera att detta förhållande av allelfrekvenser är något annorlunda än oddsförhållandet, även om de två åtgärderna konvergerar för mycket sällsynta varianter.)
ansökan till prionsjukdom
Vi gick igenom denna logik i en studie som vi publicerade tidigare i år och kvantifierade penetrans av prionsjukdomsvarianter ., Jag bryr mig om prionsjukdom av en personlig anledning — min fru har en patogen variant i PRNP — men det visar sig att prionsjukdom också är ett bra testfall för att använda ovanstående logik för att uppskatta penetrance. Ingen av individerna i ExAC v1 fastställdes på neurodegenerativ sjukdom, så ExAC är verkligen en bra populationskontroll dataset för prionsjukdom. Och eftersom prionsjukdomar är ”anmälningspliktiga”, har nationella övervakningscentrum exceptionellt bra falluppskattning, och tack vare deras generositet i att dela data kunde vi ackumulera en datauppsättning på 10.460 sekvenserade fall.,
Vi fann att>60 varianter rapporterade att orsaka prionsjukdom kumulativt har 52 alleler i ExAC. Det betyder att nästan 1 av 1000 personer har en av dessa varianter, och därmed är dessa varianter kumulativt mycket vanligare än alla prionsjukdomar (vilket orsakar ~1 i varje 5000 dödsfall), än mindre all genetisk prionsjukdom (endast ~15% av fallen är genetiska). Detta räcker för att berätta för oss att inte alla dessa varianter kan vara helt penetrerande. För att bestämma vilka varianter som var syndarna jämförde vi med fallserien., Varianter med utmärkt tidigare bevis på patogenicitet (Mendelisk segregation och musmodeller) var vanliga i fall och frånvarande från ExAC, i överensstämmelse med fullständig eller nästan fullständig penetrering. De flesta av de överskjutande allelantalet i ExAC bidrog med varianter som var ovanliga i fall och hade svaga tidigare tecken på patogenicitet — dessa varianter är förmodligen godartade eller bidrar endast med låg risk. Minst tre varianter föreföll mellanliggande, eftersom de var för vanliga i kontroller för full penetrans, men ändå berikade i fall över kontroller.,
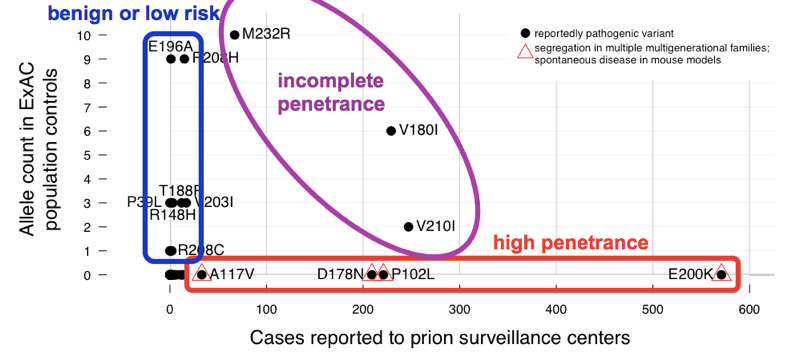
ovan: en kommenterad version av Figur 2 från .
När vi uppskattade penetrance för varje variant, med hjälp av P(D|A) formeln ovan, fann vi att det finns ett helt spektrum av penetrance för PRNP-varianter.
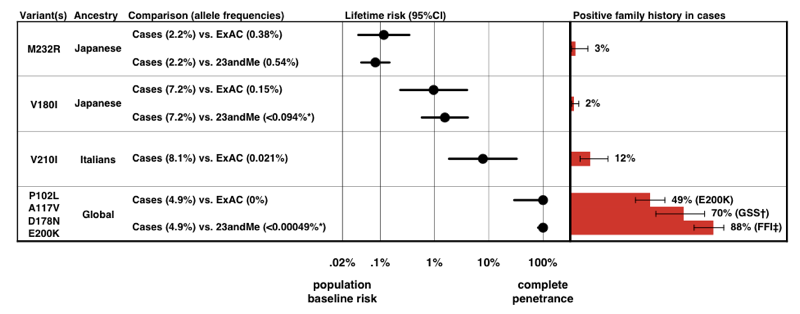
ovan: Figur 3 från .
notera skalan på X-axeln – för en sjukdom så sällsynt att den tidigare sannolikheten att utveckla den bara är 0,02%, även en 50-faldig ökning av risken är bara 1% livstidsrisk., Sammanfattningsvis uppskattar penetrance att vi härstammar från allelfrekvensinformation ensam, ganska bra med andelen fall som presenterar en positiv familjehistoria.
detta arbete har redan lett till en förändring av prognosen för vissa individer som ursprungligen hade råtts att de var i riskzonen för höggenomträngningsvarianter-se och Erika kontrollera Haydens artikel om ExAC. Du kan läsa upp min och Sonias personliga resa med denna studie här.,
ansökan till NR1H3
multipel skleros (MS) är en komplex sjukdom med många genetiska riskfaktorer , men ingen Mendelisk form av sjukdomen är känd för att existera. Tidigare i år rapporterade en studie att en missense-variant i en nukleär hormonreceptor — NR1H3 R415Q — orsakar den första någonsin Mendeliska formen av MS . Detta påstående baserades på dominerande segregering med sjukdom i två familjer, men lod-poängen var bara 2,2-under tröskeln för genomomsomfattande betydelse i familjebandstudier, vilket är mer som 3.0 eller 3.6 . Och varianten i fråga har en allelfrekvens på 0.,031% i ExAC icke-finländska européer. Det kanske inte låter som en hög allelfrekvens, men det visar sig vara alldeles för högt för denna variant för att orsaka Mendelian MS .
anser att MS har en livstidsrisk (i den allmänna befolkningen) på 0,25% hos kvinnor och 0,14% hos män . Om 0,06% av befolkningen i den allmänna befolkningen är R415Q heterozygoter, och om även hälften av dem fortsatte att utveckla MS, skulle denna variant ensam stå för 0,03% av befolkningen som utvecklar MS. så om totalt 0,25% av befolkningen utvecklar MS, bör cirka 12% av dem ha denna variant., I stället hittades varianten endast hos 1 individ av en fallserie av 2,053 MS-patienter .
detta fungerar till en allelfrekvens på 0,024% i fall, eller 0,049% om vi tillåter att 2 fall räknas i kundcaseserien. Detta är inte signifikant högre än frekvensen i ExAC. Men om denna variant orsakar MS, bör det vara vanligare i fall-mycket vanligare. Kom ihåg vår omarrangerade formel tidigare: P(D|A)/P(D) = P(A|D)/P(A). Detta innebär att om en variant ökar risken med X-fold, bör det vara X gånger vanligare i kontroller. Så om utgångsrisken för MS är 0.,25% och denna variant är 50% penetrant, den ska vara 50/.25 = 200-faldigt vanligare i fall än kontroller. Om det ens hade 10% penetrance, borde det fortfarande vara 10/.25 = 40 gånger vanligare i fall än i kontroller. Alternativt kan du tänka i termer av odds förhållanden i stället för sannolikheter. 0,25% livstidsrisk i den allmänna befolkningen betyder 1:399 odds, och om R415Q gav 50% livstidsrisk, skulle det vara 50: 50 odds. (50/50)/(1/399) = 399, så oddsförhållandet för R415Q måste vara 399 för att denna variant ska ha 50% penetrans.,
Om vi istället tillämpar vår formel med hjälp av R-koden från tidigare, förutsatt att 0.25% baseline risk och baserar beräkningen på 2 alleler på 2,053 fall, mot 21 alleler i 33,369 ExAC individer, finner vi att den övre gränsen för 95% CI på penetrance är 2.2%. Så även om R415Q var associerade med MS risk, kunde det inte ge mer än 2.2% livstidsrisk för att utveckla MS .,
i sitt formella svar och i PubMed Commons uppgav författarna en jämförelse med LRRK2 G2019S i Parkinsons sjukdom, som alla instämmer i är patogen, men som också finns i ExAC och har bara ett blygsamt oddsförhållande, beräknat till 9,6 . För den varianten fungerar matematiken. Parkinsons sjukdom är åtminstone en storleksordning vanligare än MS, med livstidsrisk uppskattas någonstans från 3.7% till 6.7%. Denna storleksordning större prevalens innebär att den ~10-faldiga anrikning som har observerats – LRRK2 G2019S finns i ungefär 0.,1% av kontrollerna och 1% av fallen-överensstämmer ungefär med den rapporterade ~ 32% livstidsrisken för Parkinsons som ges av denna variant . Dessa kvantitativa detaljer är viktiga och skiljer sig åt för varje variant och varje sjukdom. Därför är de formler som diskuteras i det här inlägget användbara, även om de bara ger mycket grova uppskattningar och är föremål för flera försiktighetsåtgärder, som förklaras nedan.
varningar
i båda de ovan beskrivna applikationerna användes allelfrekvensinformation för att få en grov uppskattning av penetrance., I prionsjukdom kunde vi visa att varianter som tidigare förutsatts mycket penetrerande gav livstidsrisk mer i storleksordningen 0.1%, 1% eller 10%. I NR1H3-historien var allelfrekvensinformation tillräcklig för att visa att den rapporterade orsaksvarianten inte kunde ge mer än några få procent livstidsrisk.
men att försöka använda allelfrekvensdata för att få en strängare uppskattning av penetrance skulle vara mycket utmanande. Till exempel har familjebaserade studier varit oense om penetrationen av PRNP E200K, med uppskattningar som sträcker sig från 60% till 90% livstidsrisk ., Sedan prion-studien kom ut har jag haft några personer från e200k-familjer frågar mig om ExAC-data kan hjälpa till att begränsa var risken ligger inom detta område. Svaret är tyvärr inte det.
Här är de viktigaste anledningarna till att jag tror att alla penetrance-uppskattningar baserade på allelfrekvens måste tas med ett saltkorn:
- om en variant är mycket penetrant, är det svårt att få en fallserie som inte innehåller relaterade individer. Om din kundcaseserie har relateds, har du tekniskt sett inte en objektiv uppskattning av P(A|D).,
- om en sjukdom är dödlig är det svårt att få en befolkningskontrollserie som inte är åtminstone något utarmad av personer med varianter som orsakar den sjukdomen. Så då har du inte heller en objektiv uppskattning av P(A).
- jämförelser av fall och kontroll allelfrekvens är sårbara för förväxling av befolkningsstratifiering. I prion-studien hade vi inte genomet-omfattande SNP-data om fall, så det fanns inget sätt att helt kontrollera detta.,
- många orsaksvarianter för sällsynta sjukdomar är så sällsynta att även med ExAC har vi ännu inte tillräckligt exakta uppskattningar av allelfrekvensen för att ge bättre än ett grovt svar.
med allt som sagt är populationsbaserad allelfrekvensuppskattning fortfarande ett bra sätt att få grova uppskattningar av penetrance i storleksordning och att utföra sanity kontroller av huruvida en genetisk variant sannolikt kan vara orsakssamband för en sällsynt sjukdom.