RNA-sekvensering (RNA-seq) har ett brett spektrum av applikationer, och det finns ingen optimal rörledning för alla fall. Vi granskar alla de viktigaste stegen i RNA-seq-dataanalys, inklusive kvalitetskontroll, läsanpassning, kvantifiering av gen-och transkriptnivåer, differentialgenuttryck, funktionell profilering och avancerad analys. De kommer att diskuteras senare.
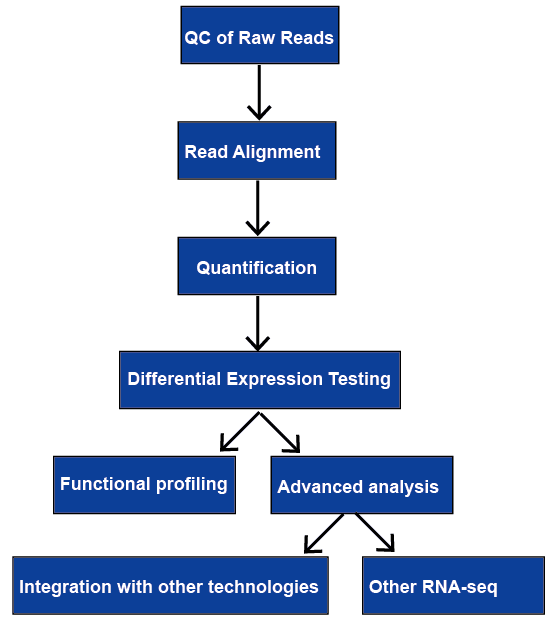
Figur 1. Det allmänna arbetsflödet av RNA-seq-analys.,
kvalitetskontroll av raw läser
kvalitetskontroll av RNA-seq raw läser består av analys av sekvenskvalitet, GC-innehåll, adapterinnehåll, överrepresenterade k-mers och duplicerade läsningar, dedikerade till att upptäcka sekvenseringsfel, föroreningar och PCR-artefakter. Läs kvalitet minskar mot 3 ’ slutet av läsningar, baser med låg kvalitet, därför bör de tas bort för att förbättra mappability., Förutom kvaliteten på rådata innehåller kvalitetskontroll av råläsningar också analys av läsanpassning (läs-enhetlighet och GC-innehåll), kvantifiering (3 ” bias, biotyper och lågt antal) och reproducerbarhet (korrelation, huvudkomponentanalys och batcheffekter).
tabell 1. Verktygen för kvalitetskontroll av RNA-seq raw läser.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Oavsett om en genom-eller transkriptomreferens är tillgänglig, kan läsningar kartlägga unikt eller tilldelas flera positioner i referensen, som kallas multi-mappade läsningar eller multireads. Genomiska multireads beror i allmänhet på repetitiva sekvenser eller delade domäner av paralogiska gener. Transcriptome Multi-mapping uppstår oftare på grund av genisoformer. Därför är transkriptidentifiering och kvantifiering viktiga utmaningar för alternativt uttryckta gener., När en referens inte är tillgänglig, RNA-seq läser monteras de novo med hjälp av paket som SOAPdenovo-Trans, oaser, Trans-ABySS eller Trinity. Pe strand-specifik och lång längd läser föredras eftersom de är mer informativa. Framväxande långläst teknik, såsom PACBIO SMRT sekvensering och Nanopore sekvensering, kan generera fullängds transkript för de flesta gener.
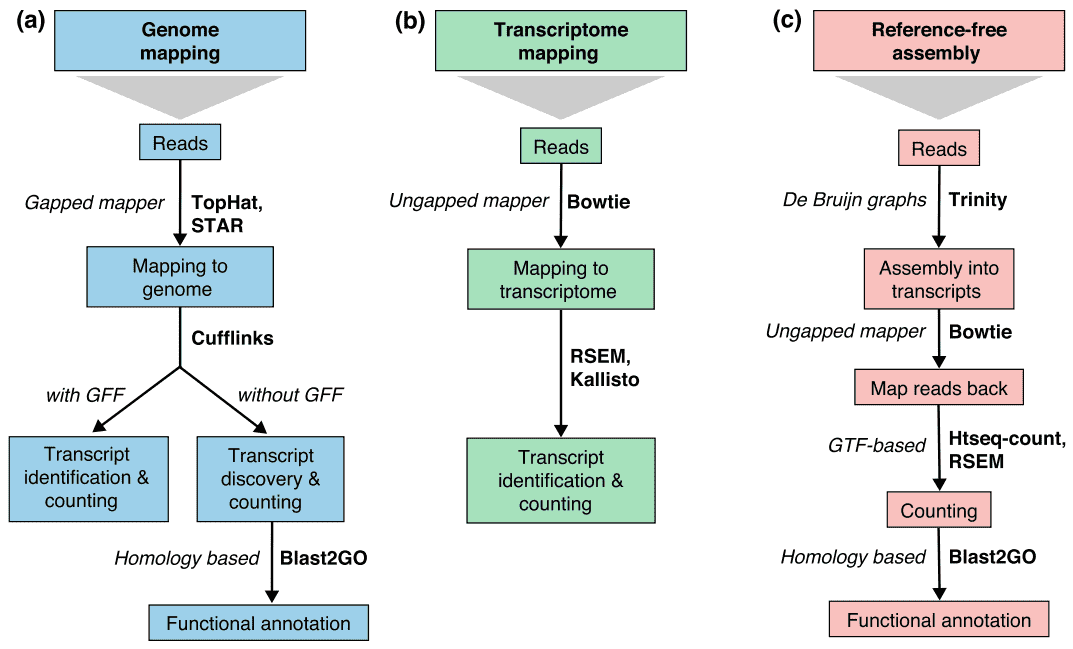
Figur 2. Tre grundläggande strategier för RNA-seq läsa kartläggning (Conesa et al. 2016)., Förkortningar: GFF, allmänt Funktionsformat; GTF, genöverföringsformat; rsem, RNA – seq genom förväntan maximering.
tabell 2. Jämförelsen av genombaserade och de novo aggregeringsstrategier för RNA-seq-analys.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| manschettknappar | utformad för att dra nytta av PE läser, och kan använda GTF-information för att identifiera uttryckta transkript, eller kan härleda transkript de novo från kartdata ensam. |
| rsem | kvantifiera uttryck från transkriptommappning. allokera Multi-mapping läser bland transkript och utgång inom-prov normaliserade värden korrigerade för sekvenseringsförskjutningar., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
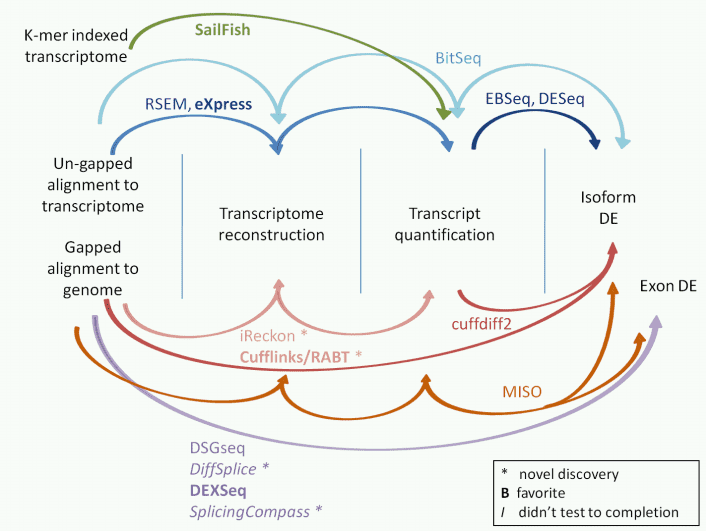
Figure 3. The tools for isoform expression quantification.,
Differential expression testing
Differential expression test används för att utvärdera om en gen är differentiellt uttryckt i ett tillstånd jämfört med den andra(s). Normaliseringsmetoder måste antas innan man jämför olika prover. RPKM och TPM normalisera bort den viktigaste faktorn, sekvenseringsdjup. TMM, DESeq och UpperQuartile kan ignorera mycket varierande och/eller starkt uttryckta funktioner., Andra faktorer som stör jämförelser mellan intra-prov innebär transkriptlängd, positionella fördomar i täckning, Genomsnittlig fragmentstorlek och GC-innehåll, som kan normaliseras med verktyg, såsom DESeq, edgeR, baySeq och NOISeq. Batcheffekter kan fortfarande vara närvarande efter normalisering, vilket kan minimeras genom lämplig experimentell design eller avlägsnas med metoder som COMBAT eller ARSyN.
tabell 5. Normaliseringsverktyg för differential expression testning.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Flera bioinformatikverktyg har utvecklats för att upptäcka från experimentella data. Jämförelsen av dessa detektionsverktyg med RNA-seq-data utfördes av Ding 2017, och resultaten visas i Tabell 7. De har visat att TopHat och dess nedströmsverktyg, Finsplice, är de snabbaste verktygen, medan PASTA är det långsammaste programmet. Dessutom kan AltEventFinder upptäcka det högsta antalet korsningar, och RSR detekterar det lägsta antalet korsningar. Andra verktyg, som TopHat, kommer sannolikt att upptäcka falska positiva., Av de två verktyg som upptäcker differentiellt skarvade isoformer, rmat är snabbare än rSeqDiff men upptäcker mindre differentiellt skarvade isoformer än rSeqDiff.
tabell 7. Detekteras som typer eller differentiellt splitsade isoformer av dessa verktyg (Ding et al. 2017).,
Visualisering
Det finns många bioinformatik verktyg för visualisering av RNA-seq data, inklusive genomet webbläsare, såsom ReadXplorer, UCSC webbläsare, Integrativ Genomik Viewer (IGV), Genomet, Savant, verktyg som är speciellt utformade för RNA-seq data, såsom RNAseqViewer, liksom vissa paket för differential genuttryck analys som möjliggör visualisering, såsom DESeq2 och DEXseq i Bioconductor. Paket, som CummeRbund och Sashimi tomter, har också utvecklats för visualisering-exklusiva ändamål.,
funktionell profilering
det senaste steget i en standard transcriptomics studie är i allmänhet karakteriseringen av de molekylära funktioner eller vägar i vilka differentiellt uttryckta gener är inblandade. Gen ontologi, Biokonduktor, DAVID eller Babelomics innehåller annoteringsdata för de flesta modellarter, som kan användas för funktionell anteckning. När det gäller nya transkript kan proteinkodande transkript funktionellt annoteras med hjälp av ortologi med hjälp av databaser som SwissProt, Pfam och InterPro., Gene ontologi (GO) möjliggör viss utbytbarhet av funktionell information över ortologer. Blast2GO är ett populärt verktyg som tillåter massiv annotering av komplett transcriptome mot en mängd olika databaser och kontrollerade vokabulärer. Rfam-databasen innehåller de flesta välkännetecknade rna-familjer som kan användas för funktionell anteckning av långa icke-kodande RNA.
avancerad analys
den avancerade analysen av RNA-seq innehåller vanligtvis andra RNA-seq och integration med annan teknik, som beskrivs i Figur 4., Mer information om tillämpningar av RNA-seq, se denna artikel tillämpningar av RNA-Seq.
Figur 3. Den avancerade analysen av RNA-seq data.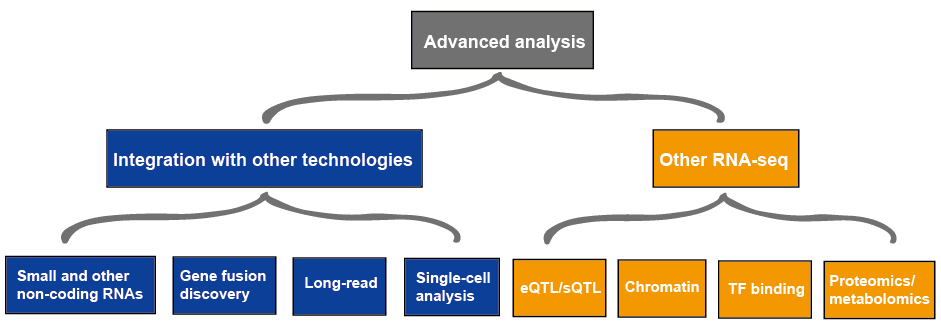
våra erfarna bioinformatikforskare är skickliga på att använda de avancerade bioinformatikverktygen för att hantera de många sekvenser som genereras av nästa och tredje generationens sekvensering. Vi erbjuder både sekvensering och bioinformatik tjänster för genomik, transcriptomics, epigenomics, mikrobiella genomik, single-cell sekvensering, och PacBio SMRT sekvensering.