ARN-secvențiere (ARN-seq) are o gamă largă de aplicații, și nu există nici o conductă optimă pentru toate cazurile. Analizăm toți pașii majori în analiza datelor ARN-seq, inclusiv controlul calității, alinierea citirii, cuantificarea genelor și a nivelurilor de transcriere, expresia genelor diferențiale, profilarea funcțională și analiza avansată. Acestea vor fi discutate mai târziu.
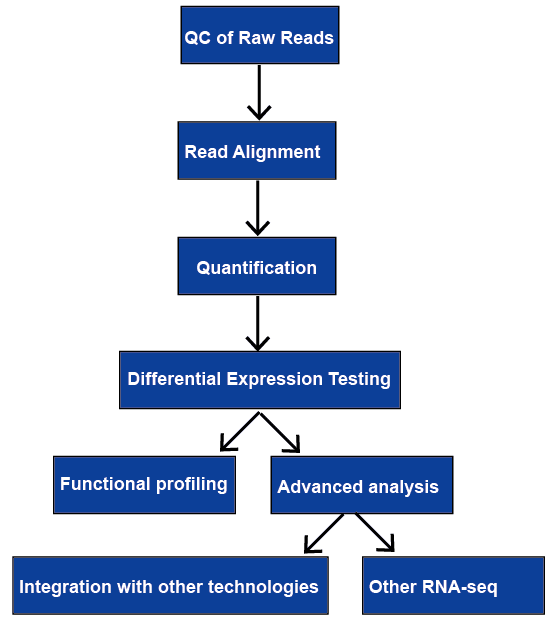
Figura 1. Fluxul general de lucru al analizei ARN-seq.,controlul calității citirilor brute controlul calității citirilor RAW ARN-seq constă în analiza calității secvenței, a conținutului GC, a conținutului adaptorului, A K-mers-urilor suprareprezentate și a citirilor duplicate, dedicate detectării erorilor de secvențiere, contaminărilor și artefactelor PCR. Calitatea citirii scade spre sfârșitul 3 ‘ al citirilor, baze cu o calitate scăzută, prin urmare, acestea ar trebui eliminate pentru a îmbunătăți mappability., În plus față de calitatea datelor brute, controlul calității citirilor brute include, de asemenea, analiza alinierii la citire (uniformitatea citirii și conținutul GC), cuantificarea (3’ biotypes și low-counts) și reproductibilitatea (corelația, analiza componentelor principale și efectele lotului).Tabelul 1. Instrumentele pentru controlul calității ARN-seq raw citește.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Indiferent dacă este disponibilă o referință genomului sau transcriptomului, citirile pot fi cartografiate în mod unic sau pot fi atribuite mai multor poziții din referință, care sunt denumite citiri sau multireade multi-mapate. Multireadele genomice se datorează, în general, secvențelor repetitive sau domeniilor partajate ale genelor paraloge. Multi-cartografierea transcriptomului apare mai des datorită izoformelor genetice. Prin urmare, identificarea transcrierii și cuantificarea sunt provocări importante pentru genele exprimate alternativ., Când o referință nu este disponibilă, citirile ARN-seq sunt asamblate de novo folosind pachete precum SOAPdenovo-Trans, Oaze, Trans-ABySS sau Trinity. Citirile specifice pe strand și de lungă durată sunt preferate, deoarece sunt mai informative. Tehnologiile emergente cu citire lungă, cum ar fi secvențierea PacBio SMRT și secvențierea Nanopore, pot genera transcrieri de lungă durată pentru majoritatea genelor.
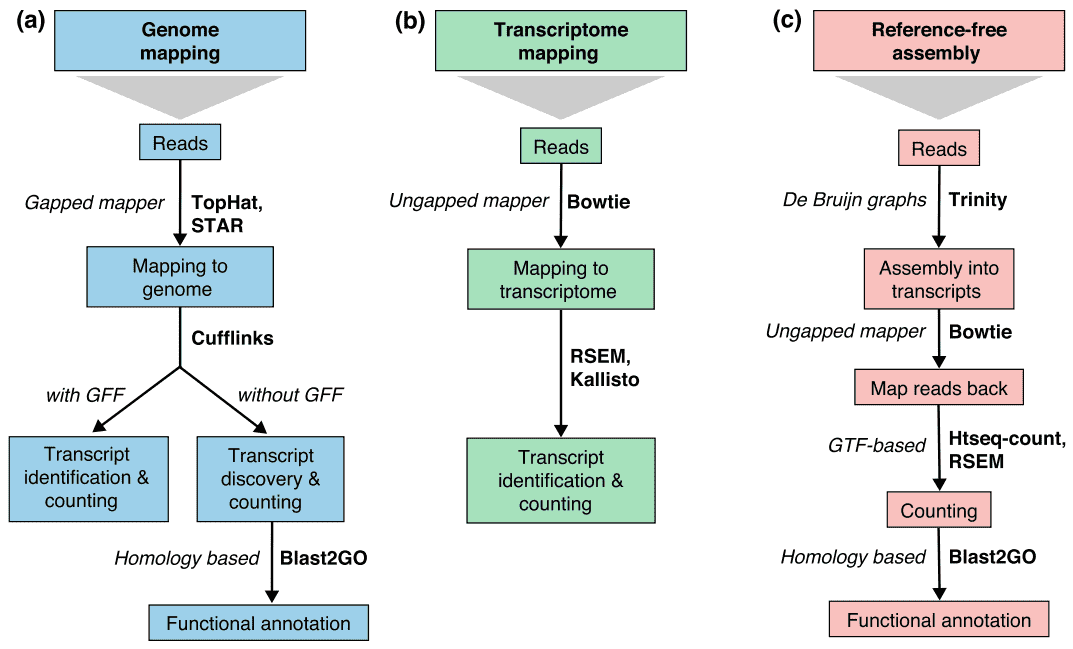
Figura 2. Trei strategii de bază pentru ARN-seq read mapping (Conesa et al. 2016)., Abrevieri: GFF, format de caracteristică generală; GTF, format de transfer de gene; RSEM, ARN-seq prin maximizarea așteptărilor.Tabelul 2. Compararea strategiilor de asamblare bazate pe genom și de novo pentru analiza ARN-seq.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Butoni | Conceput pentru a profita de PE citește, și poate folosi GTF informații pentru a identifica exprimat transcrieri, sau se poate deduce transcrieri de novo de cartografiere a datelor. |
| RSEM | Cuantifica expresie de transcriptomului de cartografiere. alocați citirile multi-mapping între transcriere și ieșire în cadrul eșantionului valori normalizate corectate pentru prejudecățile de secvențiere., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
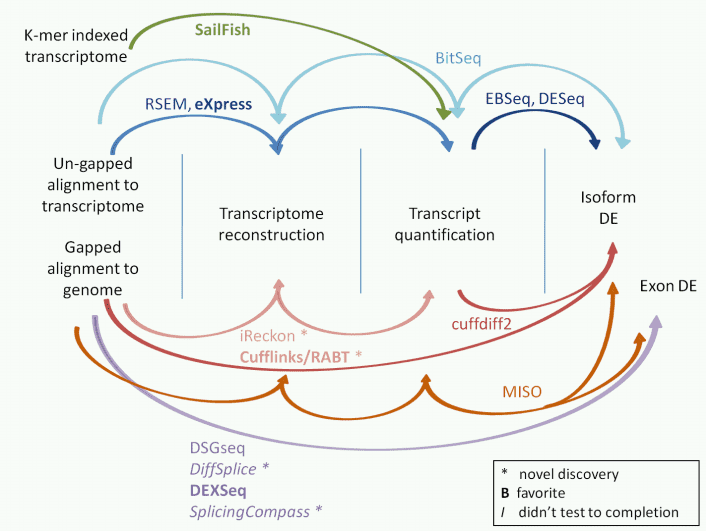
Figure 3. The tools for isoform expression quantification.,
testarea expresiei diferențiale
testarea expresiei diferențiale este utilizată pentru a evalua dacă o genă este exprimată diferențial într-o condiție în comparație cu cealaltă(celelalte). Metodele de normalizare trebuie adoptate înainte de a compara diferite probe. RPKM și TPM normalizează cel mai important factor, adâncimea de secvențiere. TMM, DESeq și UpperQuartile pot ignora caracteristicile foarte variabile și / sau foarte exprimate., Alți factori care interferează cu intra-proba comparații implica transcriere lungime, pozițional prejudecăți în acoperire medie fragment de dimensiuni și GC conținut, care poate fi normalizat de instrumente, cum ar fi DESeq, edgeR, baySeq, și NOISeq. Efectele lotului pot fi încă prezente după normalizare, care pot fi reduse la minimum prin proiectarea experimentală adecvată sau eliminate prin metode precum COMBAT sau ARSyN.Tabelul 5. Instrumentele de normalizare pentru testarea expresiei diferențiale.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Mai multe instrumente Bioinformatica au fost dezvoltate pentru a detecta ca din datele experimentale. Compararea acestor instrumente de detectare folosind date ARN-seq a fost efectuată de Ding în 2017, iar rezultatele sunt prezentate în Tabelul 7. Ei au demonstrat că TopHat și instrumentul său din aval, FineSplice, sunt cele mai rapide instrumente, în timp ce pastele sunt cel mai lent program. În plus, AltEventFinder poate detecta cel mai mare număr de intersecții, iar RSR detectează cel mai mic număr de intersecții. Alte instrumente, cum ar fi TopHat, sunt susceptibile de a detecta cele fals pozitive., De două instrumente care detecta diferențiat îmbinat izoforme, rMATS este mai rapid decât rSeqDiff dar detectează mai puțin diferențiat îmbinat izoforme decât rSeqDiff.Tabelul 7. Detectate ca tipuri sau izoforme diferențiate ale acestor instrumente (Ding și colab . 2017).,
Vizualizare
Există multe bioinformatica instrumente pentru vizualizarea de ARN-următoarele date, inclusiv genomul browsere, cum ar fi ReadXplorer, UCSC browser, Integratoare Genomica Viewer (IGV), Genomul Hărți, Savant, instrumente special concepute pentru ARN-următoarele date, cum ar fi RNAseqViewer, precum și unele pachete pentru diferențial expresia genelor analiză care să permită vizualizarea, cum ar fi DESeq2 și DEXseq în Bioconductor. Pachetele, cum ar fi parcelele CummeRbund și Sashimi, au fost, de asemenea, dezvoltate în scopuri exclusiv de vizualizare.,cel mai recent pas dintr-un studiu standard de transcriptomică este, în general, caracterizarea funcțiilor moleculare sau a căilor în care sunt implicate genele exprimate diferențiat. Ontologia genelor, Bioconductorul, DAVID sau Babelomica conțin date de adnotare pentru majoritatea speciilor de model, care pot fi utilizate pentru adnotarea funcțională. Ca pentru roman transcrieri, proteine-codare transcrierile pot fi din punct de vedere funcțional adnotat folosind orthology cu ajutorul bazelor de date, cum ar fi SwissProt, Pfam, și InterPro., Ontologia genetică (GO) permite o anumită schimbabilitate a informațiilor funcționale în ortologii. Blast2GO este un instrument popular care permite adnotarea masivă a transcriptomului complet împotriva unei varietăți de baze de date și vocabulare controlate. Baza de date Rfam conține cele mai bine caracterizate familii de ARN care pot fi utilizate pentru adnotarea funcțională a ARN-urilor lungi care nu codifică.analiza avansată analiza avansată a ARN-seq include de obicei alte ARN-seq și integrarea cu alte tehnologii, care este prezentată în Figura 4., Mai multe informații despre aplicații de ARN-seq, vă rugăm să consultați acest articol aplicații de ARN-Seq.figura 3. Analiza avansată a datelor ARN-seq.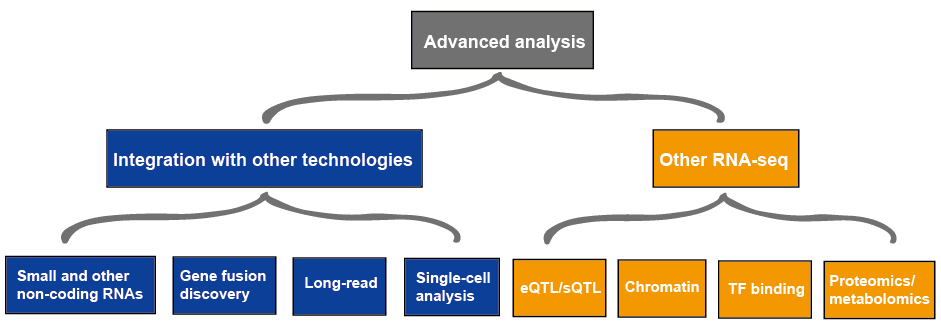
Nostru experimentat bioinformatica oamenii de stiinta sunt calificați în utilizarea avansată bioinformatica instrumente pentru a face față cu numeroase secvențe generate de următoarea și a treia generație de secvențiere. Oferim servicii de secvențiere și bioinformatică pentru genomică, transcriptomică, epigenomică, genomică microbiană, secvențiere cu o singură celulă și secvențiere PacBio SMRT.