RNA-Seq (RNA-seq) ma szeroki zakres zastosowań i nie ma optymalnego rurociągu dla wszystkich przypadków. Przeglądamy wszystkie główne etapy analizy danych RNA-seq, w tym kontrolę jakości, wyrównanie odczytu, kwantyfikację poziomów genów i transkrypcji, ekspresję różnicową genów, profilowanie funkcjonalne i zaawansowaną analizę. Omówimy je później.
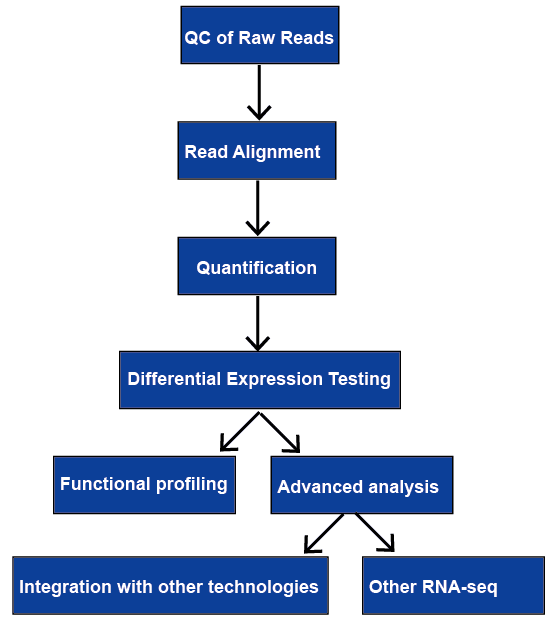
Rysunek 1. Ogólny przepływ pracy analizy RNA-seq.,
Kontrola jakości odczytów surowych
Kontrola jakości odczytów surowych RNA-seq składa się z analizy jakości sekwencji, zawartości GC, zawartości adaptera, nadreprezentowanych k-merów i duplikowanych odczytów, dedykowanych do wykrywania błędów sekwencjonowania, zanieczyszczeń i artefaktów PCR. Jakość odczytu spada w kierunku 3 ' końca odczytu, bazy o niskiej jakości, dlatego należy je usunąć, aby poprawić mapowalność., Oprócz jakości danych surowych, kontrola jakości odczytów surowych obejmuje również analizę wyrównania odczytu( jednolitość odczytu i zawartość GC), kwantyfikację (odchylenie 3′, biotypy i niskie liczby) i odtwarzalność (korelacja, analiza głównych składników i efekty partii).
Tabela 1. Narzędzia do kontroli jakości odczytów RNA-seq raw.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Niezależnie od tego, czy odniesienie do genomu lub transkryptomu jest dostępne, odczyty mogą być mapowane jednoznacznie lub przypisane do wielu pozycji w referencji, które są określane jako odczyty wielomapowane lub wielowarstwowe. Multiready genomowe są na ogół spowodowane powtarzającymi się sekwencjami lub współdzielonymi domenami genów paralogicznych. Multiplikacja transkryptomu powstaje częściej z powodu izoform genów. Dlatego identyfikacja transkryptu i kwantyfikacja są ważnymi wyzwaniami dla genów ekspresji alternatywnej., Gdy Referencja nie jest dostępna, odczyty RNA-seq są montowane de novo za pomocą pakietów takich jak SOAPdenovo-Trans, Oases, Trans-ABySS lub Trinity. PE strand-specific I long-length czyta są preferowane, ponieważ są one bardziej pouczające. Powstające długo czytane technologie, takie jak sekwencjonowanie PacBio SMRT i sekwencjonowanie Nanopore, mogą generować transkrypty pełnej długości dla większości genów.
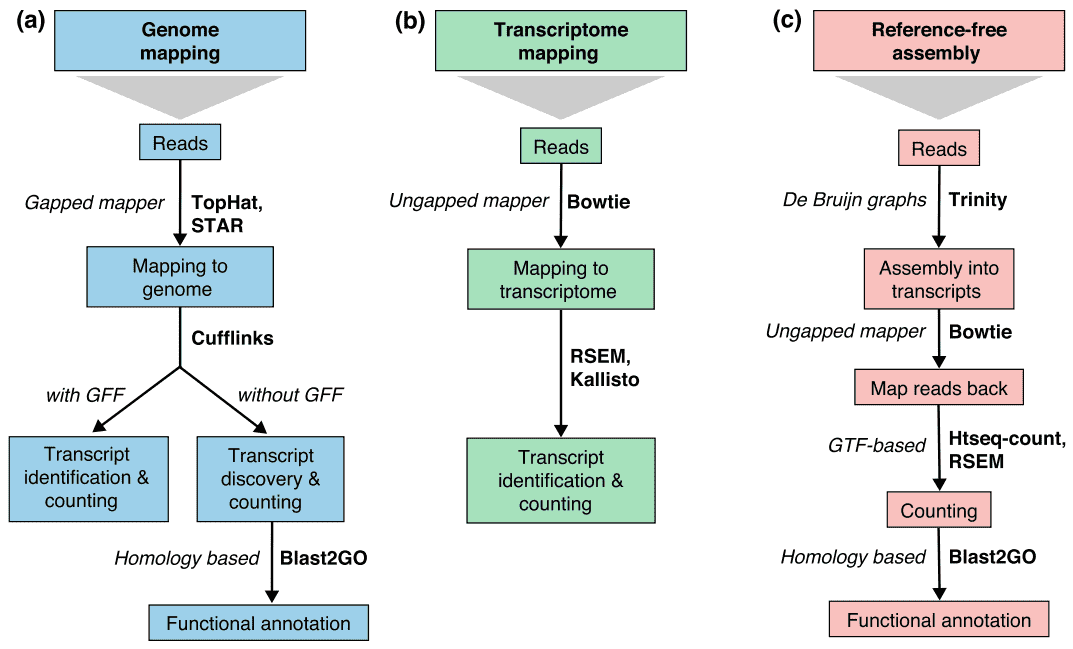
Rysunek 2. Trzy podstawowe strategie mapowania odczytu RNA-seq (Conesa et al. 2016)., Skróty: GFF, ogólny format funkcji; GTF, format transferu genów; RSEM, RNA-seq przez maksymalizację oczekiwań.
Tabela 2. Porównanie strategii montażu opartych na genomie i De novo dla analizy RNA-seq.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Spinki Do Mankietów | zaprojektowane do korzystania z odczytów PE i mogą wykorzystywać informacje GTF do identyfikacji wyrażonych transkryptów lub mogą wywnioskować transkrypty de novo z samych danych mapowania. |
| RSEM | Oblicz wyrażenie z mapowania transkryptomu. Przydziel odczyty wielomapowe między transkrypcjami i wyjściowymi wartościami znormalizowanymi w próbce skorygowanymi o błędy sekwencjonowania., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
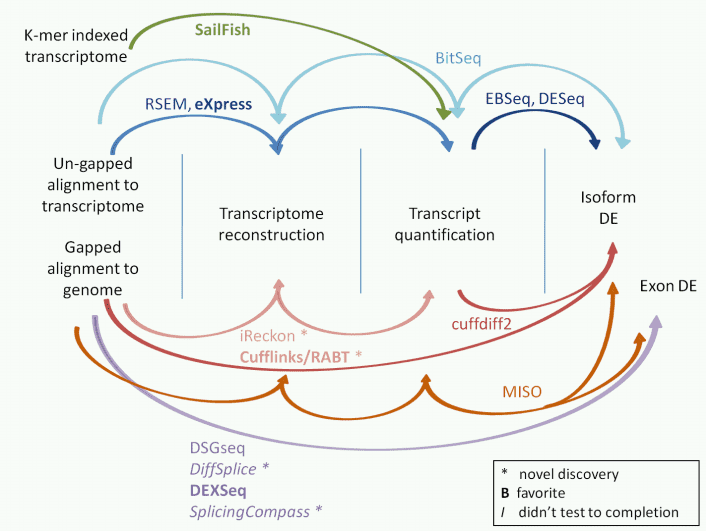
Figure 3. The tools for isoform expression quantification.,
testowanie ekspresji różnicowej
testowanie ekspresji różnicowej służy do oceny, czy jeden gen jest różnie wyrażony w jednym stanie w porównaniu do drugiego(s). Metody normalizacji muszą być przyjęte przed porównaniem różnych próbek. RPKM i TPM normalizują najważniejszy czynnik, czyli głębokość sekwencjonowania. TMM, DESeq i UpperQuartile mogą ignorować wysoce zmienne i / lub wysoce wyrażone funkcje., Inne czynniki, które zakłócają porównania wewnątrz próbki obejmują długość transkrypcji, błędy pozycyjne w zasięgu, średni rozmiar fragmentu i zawartość GC, które mogą być znormalizowane przez narzędzia, takie jak DESeq, edgeR, baySeq i NOISeq. Efekty partii mogą być nadal obecne po normalizacji, które mogą być zminimalizowane przez odpowiedni projekt eksperymentalny, lub usunięte za pomocą metod takich jak walka lub ARSyN.
Tabela 5. Narzędzia normalizacyjne do testowania ekspresji różniczkowej.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Opracowano wiele narzędzi bioinformatycznych do wykrywania danych eksperymentalnych. Porównanie tych narzędzi wykrywania z wykorzystaniem danych RNA-seq zostało przeprowadzone przez Ding w 2017 roku, a wyniki przedstawiono w tabeli 7. Wykazały one, że TopHat i jego narzędzie finesplice są najszybszymi narzędziami, podczas gdy PASTA jest najwolniejszym programem. Ponadto AltEventFinder może wykryć największą liczbę skrzyżowań, a RSR wykrywa najniższą liczbę skrzyżowań. Inne narzędzia, takie jak TopHat, mogą wykrywać fałszywie dodatnie., Z dwóch narzędzi, które wykrywają różne izoformy splicated, rMATS jest szybszy niż rSeqDiff, ale wykrywa mniej izoform splicated differentially niż rSeqDiff.
Tabela 7. Wykryto jako typy lub różnie splatane izoformy tych narzędzi (Ding et al. 2017).,
Wizualizacja
istnieje wiele narzędzi bioinformatycznych do wizualizacji danych RNA-seq, w tym przeglądarki genomów, takie jak ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Mapy genomu, Savant, narzędzia specjalnie zaprojektowane do danych RNA-seq, takie jak RNAseqViewer, a także niektóre pakiety do analizy ekspresji genów różnicowych, które umożliwiają wizualizację, takie jak DESeq2 i DEXseq w Bioconductor. Pakiety, takie jak CummeRbund i sashimi, zostały również opracowane w celach wyłącznie wizualizacyjnych.,
Profilowanie funkcjonalne
ostatnim krokiem w standardowym badaniu transkryptomicznym jest ogólnie charakterystyka funkcji molekularnych lub szlaków, w których zaangażowane są geny o różnej ekspresji. Ontologia genów, Bioconductor, DAVID, czy Babelomics zawierają dane adnotacji dla większości gatunków modelowych, które mogą być wykorzystane do adnotacji funkcjonalnej. Podobnie jak w przypadku nowych transkryptów, transkrypty kodujące białka mogą być funkcjonalnie adnotowane za pomocą ortologii za pomocą baz danych takich jak SwissProt, Pfam i InterPro., Ontologia genów (GO) pozwala na pewną wymianę informacji funkcjonalnych między ortologami. Blast2GO jest popularnym narzędziem, które umożliwia masową adnotację kompletnego transkryptomu przeciwko wielu bazom danych i kontrolowanym słownikom. Baza danych Rfam zawiera najbardziej dobrze scharakteryzowane rodziny RNA, które mogą być użyte do funkcjonalnej adnotacji długich niekodujących RNA.
zaawansowana analiza
zaawansowana analiza RNA-seq zwykle obejmuje inne RNA-seq i integrację z innymi technologiami, co przedstawiono na rysunku 4., Więcej informacji na temat zastosowań RNA-seq można znaleźć w artykule zastosowania RNA-Seq.
Rysunek 3. Zaawansowana analiza danych RNA-seq.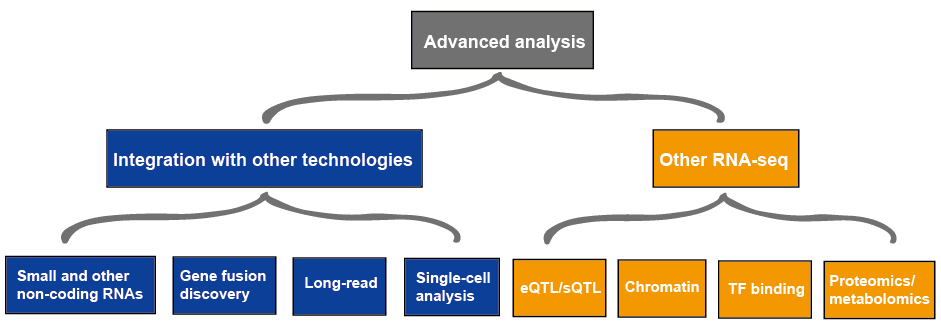
nasi doświadczeni naukowcy bioinformatycy są wykwalifikowani w wykorzystaniu zaawansowanych narzędzi bioinformatycznych do radzenia sobie z licznymi sekwencjami generowanymi przez sekwencjonowanie następnej i trzeciej generacji. Świadczymy zarówno usługi sekwencjonowania i bioinformatyki dla genomiki, transkryptomiki, epigenomiki, genomiki drobnoustrojów, sekwencjonowania jednokomórkowego i sekwencjonowania PacBio SMRT.