vorige maand was ik uitgenodigd om te spreken op de Centers for Mendelian Genomics (CMG) Analysis and Methods Development meeting over “Population-based estimation of penetrantie in rare disease”. Hier is de blog post versie van mijn talk.
Wat is penetrantie en waarom kan het ons schelen?
penetrantie is de kans op het ontwikkelen van een bepaalde ziekte met een bepaald genotype., Men kan spreken van leeftijdsafhankelijke penetrantie, dus het percentage mensen met een genotype dat de ziekte ontwikkelt op 40-jarige leeftijd, op 50-jarige leeftijd, enzovoort; ik spreek meestal in termen van levenslange risico ‘ s, dat wil zeggen de kans dat je ooit de ziekte ontwikkelt voordat je sterft. Inherent hieraan is dat, Voor ziekten die bij volwassenen ontstaan, levenslange risico ‘ s nooit helemaal 100% kunnen zijn, omdat je altijd eerst aan iets anders zou kunnen sterven.
penetrantie is enorm belangrijk voor individuen die voorspellende genetische tests ondergaan — de eerste vraag van veel mensen is: “betekent dit dat ik de ziekte zeker zal krijgen?”., Toch is het vaak erg moeilijk om te komen door een stevige schatting van penetrantie.
traditionele methoden voor het schatten van penetrantie
in een ideale wereld zou de juiste manier om penetrantie te schatten zijn om vanaf de geboorte een grote cohort van mensen met een bepaald genotype vast te stellen, ze te volgen totdat ze allemaal aan iets of iets anders zijn gestorven, en dan te vragen hoeveel de ziekte ooit hebben ontwikkeld voordat ze stierven. Sinds genotypering technologie werd uitgevonden minder dan een mensenleven geleden, is dit nooit gedaan voor een ziekte.
in plaats daarvan gebruiken onderzoekers vaak familiegebaseerde methoden om penetrantie te schatten., Een typische studie zou kijken naar iedereen die is waargenomen met het gegeven genotype, en vragen hoeveel hebben diease, of hoeveel hebben ziekte op een bepaalde leeftijd. Familie-gebaseerde methoden lijden aan doordringende vaststelling bias ., de families die oorspronkelijk werden gebruikt om vast te stellen dat de variant de ziekte veroorzaakt, zijn opgenomen in de analyse
als voorbeeld van dit laatste punt, bij genetische prionziekte kiest slechts 23% van de mensen die risico lopen voor voorspellende genetische tests , en in stamboomgegevens die ik heb gehad aan, we kenden de genotypes van slechts 22% van de risico ‘ s individuen .,
alle hierboven genoemde factoren werken in dezelfde richting en hebben de neiging om de penetrantieschatting te vergroten.
onderzoekers zijn zich al lange tijd bewust van deze problemen en hebben enkele oplossingen voorgesteld. Als één voorbeeld, impliceert de kin-cohortmethode het vaststellen van gezonde individuen willekeurig van een bevolking, genotypering hen, het nemen van een familiegeschiedenis, en het vergelijken van overlevingscurven van hun eerstegraads verwanten., Dit is een zeer slimme oplossing, maar het is afhankelijk van de mogelijkheid om een groot genoeg aantal mensen met een ziekteverwekkende genotype vast te stellen zonder vast te stellen op de aanwezigheid van de ziekte. Het werkte dus voor BRCA1-en BRCA2-varianten in Amerikaanse Ashkenazische Joden, maar voor veel zeldzamere genetische aandoeningen is het onpraktisch, omdat je tientallen of honderdduizenden mensen zou moeten rekruteren om zelfs maar één individu met een genotype van interesse te vinden.,
population-based methods
om alle hierboven beschreven redenen is het zeer nuttig om orthogonale, population-based methods te hebben voor het stellen van vragen over penetrantie. Het eerste belangrijke inzicht hier is dat een volledig penetrant genetische variant niet gemeenschappelijker in de bevolking zou moeten zijn dan de ziekte die het veroorzaakt. Het toepassen van deze logica in de praktijk betekent dat je goede schattingen van allelfrequentie nodig hebt, zelfs voor ongewone varianten, en dat is moeilijk om aan te komen tot voor kort. ExAC, een database van genetische variatie in 60.706 menselijke exomes, biedt nieuwe kansen ., Veel individuen in ExAC werden vastgesteld als gevallen of controles voor verschillende gemeenschappelijke, complexe ziekten, maar geen werden vastgesteld voor Mendeliaanse ziekte, dus ExAC is een goede referentiedatabase voor het bestuderen van de meeste genetische ziekten.door informatie over de allelfrequentie in de algemene populatie te verstrekken, heeft ExAC, net als eerdere referentiedatabases zoals ESP , duidelijk gemaakt dat klinische genetica een groot probleem heeft: veel varianten waarvan is gemeld dat ze genetische ziekte veroorzaken, veroorzaken eigenlijk geen genetische ziekte, of in ieder geval meestal niet.,
twee databases-HGMD en ClinVar-verzamelen beweringen uit de literatuur en uit klinische laboratoria waarin wordt gesteld dat een bepaalde genetische variant een bepaalde genetische ziekte veroorzaakt. Bij de laatste telling, waren er meer dan 100.000 unieke gerapporteerde ziekte-veroorzakende genetische varianten in deze databases. De gemiddelde persoon in ExAC heeft 54 van hen . Het is duidelijk dat de gemiddelde persoon geen 54 genetische ziekten heeft., Natuurlijk, veel van deze overmaat wordt veroorzaakt door een klein aantal Wild hoogfrequente varianten die duidelijk geen genetische ziekte veroorzaken, en veel van het kan zijn naar verluidt recessieve varianten gevonden in een heterozygote staat in ExAC. Maar zelfs als we kijken naar varianten in dominante ziektegenen met een allelfrequentie van <1%, zien we nog steeds 0,89 naar verluidt pathogene varianten per persoon, en het is duidelijk niet het geval dat ~ 90% van de mensen een dominante genetische ziekte hebben., Over het allelfrequentiespectrum heen zijn er veel naar verluidt pathogene varianten die niet zo pathogeen zijn. Toen Anne O ‘ Donnell en ik naar de naar verluidt pathogene varianten met de hoogste allelfrequenties in ExAC keken, en vroegen hoe ze er in geslaagd waren om verkeerd geclassificeerd te worden als pathogeen, ontdekten we dat het probleem meestal terug te voeren was op een paper in de literatuur die een claim van pathogeniteit had gemaakt op basis van onvoldoende bewijs.
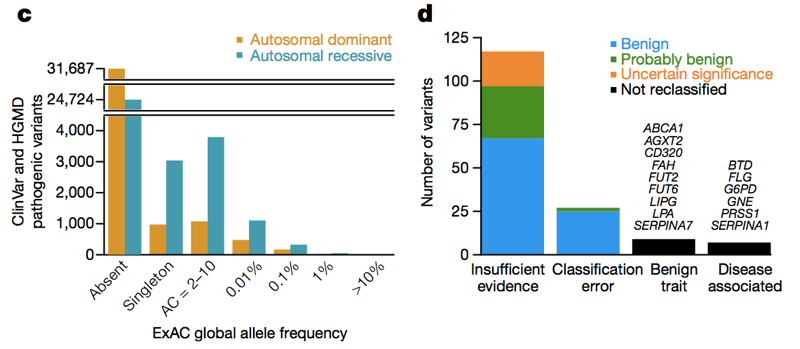
hierboven: figuren 3C en 3D uit ., Over het allelfrequentiespectrum en in zowel dominante als recessieve ziektegenen, zijn er veel naar verluidt pathogene varianten die in ExAC voorkomen. Van hoge (>1%) frequentie naar verluidt pathogene varianten, enkele zijn echt pathogeen, sommige zijn echt eigenschap-geassocieerd, maar de eigenschap is goedaardig, en sommige zijn annotatiefouten in databases — maar de meeste zijn gebaseerd op literatuur met onvoldoende bewijs.,dankzij de informatie van ExAC over de Allelfrequentie konden nu meer dan 200 genetische varianten worden herclassificeerd van pathogeen naar goedaardig, waarschijnlijk goedaardig of van onzekere significantie . Dit soort herindelingen leiden soms tot terugslag van de oorspronkelijke auteurs die stelden dat een variant een genetische ziekte veroorzaakt, die kunnen beweren dat een variant nog steeds pathogeen zou kunnen zijn, maar met onvolledige penetrantie. Maar hoe “incompleet” kan onvolledige penetrantie zijn?, We moeten kwantitatief te krijgen, want als levenslange risico is maximaal 1%, dan is het nog steeds redelijk om te zeggen dat een variant “veroorzaakt” een genetische ziekte, of is “pathogeen”? Hoewel allelfrequentie-informatie nooit kan bewijzen dat een variant geen verband heeft met ziekte, kan zij grenzen stellen aan wat de mogelijke penetrantie zou kunnen zijn, en in veel gevallen, zelfs voor vrij zeldzame varianten, is het mogelijk om aan te tonen dat er geen manier is dat een variant een risiconiveau geeft ergens in de buurt van 100%.,
om kwantitatief te krijgen, moeten we onze eerdere observatie uitbreiden — dat een volledig penetrerende genetische variant niet vaker voorkomt in de populatie dan de ziekte die het veroorzaakt. Dit is allemaal eenvoudige wiskunde en populatiegenetica, maar het wordt te vaak niet toegepast in de praktijk. Hier zijn twee manieren waarop we kunnen denken over allelfrequentie bij het maken van gevolgtrekkingen over pathogeniteit en penetrantie.
maximale geloofwaardige allelfrequentie
stel dat u het exoom van een patiënt met Mendeliaanse ziekte bestudeert en probeert de causale variant te identificeren., Mijn collega James Ware heeft een strategie bedacht om dat exoom te filteren tegen de allelfrequentie informatie in ExAC, gebruikmakend van de volgende logica., De maximale allel frequentie dat aannemelijk is voor een variant van de oorzaak van een dominant erfelijke ziekte is gelijk aan de prevalentie van de ziekte keer de allelen van heterogeniteit (deel van de gevallen toe te schrijven aan een variant) gedeeld door penetrantie (minder penetrant varianten kan worden vaker), gedeeld door 2 (want we zijn diploïd):
\
bijvoorbeeld, prion ziekte veroorzaakt bij 1 op de 5.000 sterfgevallen, en de meest voorkomende variant (E200K) is te vinden in 5% van de gevallen , dus een 100% penetrant variant niet beschikken allel frequentie van meer dan 0.0005% (1 op de 200.000) ., Cardiomyopathie komt voor bij 1 op de 500 mensen, de meest voorkomende variant wordt gevonden in <2% van de gevallen, dus een penetrant variant van 50% kan geen allelfrequentie hebben van meer dan 0,004% . De formule voor recessieve ziekten is een inkeping ingewikkelder maar James heeft het ook uitgewerkt en het wordt beschreven in .terwijl mensen in het verleden vaak varianten met een allelfrequentie hebben uitgefilterd >0,1% wanneer ze proberen de oorzaak van een dominante ziekte te identificeren , kunnen we eigenlijk veel strenger zijn., Het voorbehoud is dat bij lage allel tellingen, ons vermogen om de allelfrequentie te schatten wordt beperkt door steekproefvariantie. Als we bijvoorbeeld kijken naar varianten die gezien worden met een allelfrequentie van 1% bij Europeanen in ESP, dan hebben deze varianten ook een frequentie van ongeveer 1% bij ExAC Europeanen. Maar varianten met een frequentie van 0,1% in ESP neigen iets zeldzamer te zijn in ExAC, en de meeste singletons (varianten die precies één keer in ESP worden gezien) verschijnen niet een tweede keer in ExAC.
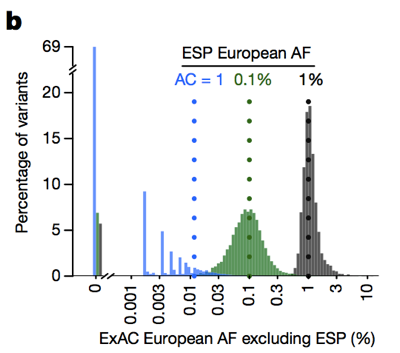
hierboven: figuur 3B vanaf . Hoe lager het allelgetal, hoe minder goed een schatting van de allelfrequentie die het biedt.,
daarom, hoe lager het allelaantal, hoe conservatiever we moeten zijn. We hebben een raamwerk bedacht om dit te doen met behulp van de 95% bovengrens van de poisson distributie op hoeveel allelen kunnen worden waargenomen bij een bepaalde frequentie, en hebben vooraf berekende waarden voor alle ExAC (beschikbaar op FTP) die u kunt gebruiken-Lees meer over de methoden in . James heeft ook deze handige web app die u toelaat om te verkennen wat de “maximale geloofwaardige allelfrequentie” moet zijn voor uw ziekte van belang.,
Inherent aan deze benadering is dat hoe lager de penetrantie van een variant, hoe hoger de frequentie kan zijn bij de algemene bevolking. Maar je moet ook bedenken dat als de penetrantie vrij laag is, bijvoorbeeld minder dan 10%, dan is het klinische nut van die variant ook laag. James en Nicky Whiffin hebben gegevens gepresenteerd om aan te tonen dat bijna alle klinische bruikbaarheid van sequencing in cardiomyopathie afkomstig is van varianten met een frequentie van <0,001% — meer voorkomende varianten dragen cumulatief weinig of geen risico bij .,
schatting en grenzen van het levenslange risico
onthoud dat penetrantie de kans op ziekte is bij een bepaald genotype. Of, als we kijken naar een allelisch in plaats van genotypisch model, de kans op ziekte gegeven een bepaald allel. We kunnen dit schrijven als P (D / A). Zodra we dat doen, wordt het duidelijk dat, volgens de stelling van Bayes,
\
elk van deze termen een bepaalde betekenis heeft:
merk hier op dat “population controls” een groep betekent die niet geselecteerd is voor de aanwezigheid, noch voor de afwezigheid van de ziekte. Slechts een deel van de bevolking.
So:
\
deze logica is niets nieuws., Het gebruik van de stelling van Bayes om het risico van ziekten te schatten dateert minstens terug tot de schatting van het risico van kanker in rokers , en de toepassing ervan op genetica is bijna even lang overwogen . Maar om deze vergelijking te laten werken voor zeldzame ziekten, heb je vrij goede schattingen nodig van de allelfrequentie van gevallen en populaties, en die waren tot voor kort moeilijk te verkrijgen. Dus dankzij ExAC zijn er steeds meer situaties waarin deze vergelijking relevant is.
Hier is de R-code die ik heb geschreven (oorspronkelijk hier) om penetrantie te schatten op basis van deze formule.,
Als u de R-code niet zelf wilt uitvoeren, heeft James Ware het geà mplementeerd in het tabblad “penetrrance” van deze web-app, zodat u gewoon uw nummers in uw browser kunt aansluiten.
om de 95% betrouwbaarheidsintervallen op penetrantie te schatten, heb ik de benadering van . U voert de alleltelling (AC) en het aantal personen (N) in voor gevallen en controles, en de bovengrens van de 95% – BI wordt berekend op basis van de bovenste 95% – BI van de binomiale verdeling voor gevallen-allelfrequentie en de onderste 95% – BI voor controles., Omgekeerd is de ondergrens van penetrantie gebaseerd op de ondergrens van de casus allelfrequentie en de bovengrens van de regel allelfrequentie. U kunt terecht kibbelen dat omdat deze formule 95% CIs gebruikt op beide allelfrequentie-waarden, de resulterende betrouwbaarheidsintervallen groter zijn dan ze zouden moeten zijn. Je zou ook terecht kunnen kibbelen dat de binomiale verdeling geen goede schatter is bij lage allel tellingen, vanwege de bias geà llustreerd in Figuur 3B hierboven getoond (en ik zou zeker nooit deze formule toepassen op singletons — varianten die slechts één keer in ExAC worden waargenomen)., Maar aan het eind van de dag, om redenen die Ik zal bespreken dichter bij het einde van deze post, deze formule is echt het beste gebruikt voor het verkrijgen van een ballpark, orde van grootte schatting van penetrantie. Als u op zoek bent naar een uiterst precieze punt schatting van penetrantie, deze hele aanpak zal waarschijnlijk niet werken voor u toch.
Als u de vergelijking herschikt, is een andere manier om er over na te denken:
\
Dit betekent dat het verhoogde risico bij mensen met een genotype evenredig is met de verhouding tussen de allelfrequentie van het geval en de populatiecontrole., Dus een variant die het risico met 200-voudig verhoogt zou 200 keer vaker voor moeten komen bij de gevallen dan bij de algemene bevolking. (Merk op dat deze verhouding van allelfrequenties iets anders is dan odds ratio hoewel de twee maten convergeren voor zeer zeldzame varianten.)
toepassing op prionziekte
deze logica hebben we gevolgd in een eerder dit jaar gepubliceerde studie, waarin de penetrantie van prionziektevarianten werd gekwantificeerd ., Ik geef om prionziekte om een persoonlijke reden-mijn vrouw herbergt een pathogene variant in PRNP — maar het blijkt dat prionziekte ook een goede testcase is voor het gebruik van de bovenstaande logica om penetrantie te schatten. Geen van de individuen in ExAC v1 werden vastgesteld op neurodegeneratieve ziekte, dus ExAC is echt een goede populatie controle dataset voor prion ziekte. En omdat prionziekten “meldplichtig” zijn, hebben nationale surveillancecentra uitzonderlijk goede gevallen vastgesteld, en dankzij hun vrijgevigheid in het delen van gegevens, waren we in staat om een dataset van 10.460 opeenvolgende gevallen te verzamelen.,
We vonden dat de >60 varianten die gezamenlijk prionziekte veroorzaken 52 allelen hebben in ExAC. Dat betekent dat bijna 1 op de 1.000 mensen een van deze varianten heeft, en dus komen deze varianten cumulatief veel vaker voor dan alle prionziekte (die ~1 op de 5.000 sterfgevallen veroorzaakt), laat staan alle genetische prionziekte (slechts ~15% van de gevallen is genetisch). Dit is genoeg om ons te vertellen dat niet al deze varianten mogelijk volledig penetrant kunnen zijn. Om te bepalen welke varianten de schuldigen waren, hebben we vergeleken met de caseserie., Varianten met uitstekende voorafgaande bewijs van pathogeniteit (Mendeliaanse segregatie en muismodellen) waren gebruikelijk in gevallen en Afwezig van ExAC, consistent met volledige of bijna volledige penetrantie. Het grootste deel van de exac — alleltelling werd bijgedragen door varianten die in gevallen ongewoon waren en die eerder zwak bewijs van pathogeniteit hadden-deze varianten zijn waarschijnlijk goedaardig of dragen slechts een laag risico bij. Ten minste drie varianten bleken tussendoor te komen, omdat ze te veel voorkwamen bij controles voor volledige penetrantie, maar nog steeds verrijkt in gevallen boven controles.,
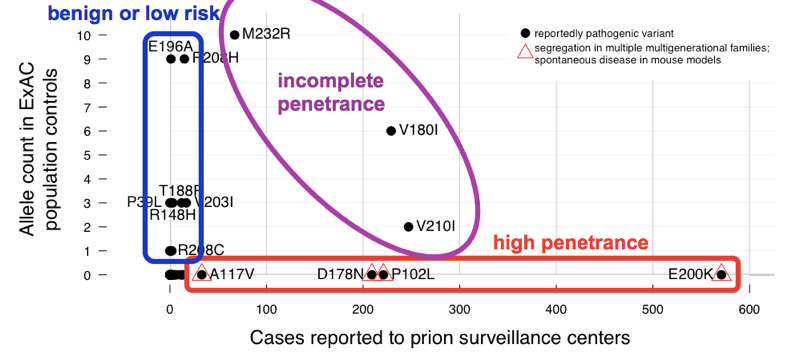
hierboven: een geannoteerde versie van Figuur 2 uit .
toen we de penetrantie voor elke variant schatten, met behulp van de P(D|A) formule hierboven, vonden we dat er een heel spectrum van penetrantie is voor PRNP-varianten.
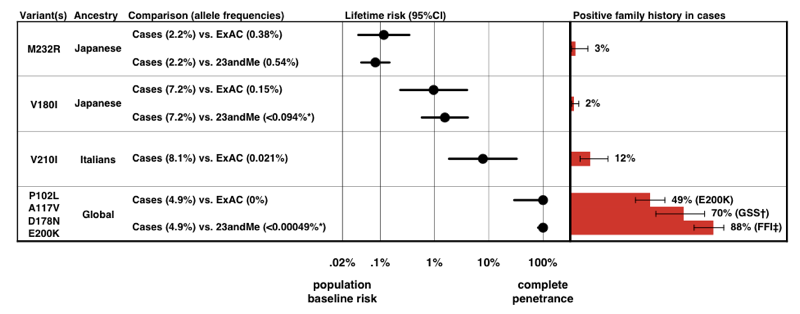
hierboven: Figuur 3 van .
noteer de schaal op de x-as-voor een ziekte die zo zeldzaam is dat de eerdere kans op ontwikkeling slechts 0,02% is, zelfs een 50-voudige toename in risico is slechts 1% levenslange risico., Geruststellend, de penetrantie schattingen dat we afleiden uit allelfrequentie informatie alleen overeen heel goed met het aandeel van de gevallen die aanwezig zijn met een positieve familiegeschiedenis.
Dit werk heeft al geleid tot een verandering in de prognose voor sommige personen die oorspronkelijk waren geadviseerd dat zij risico liepen op varianten met een hoge penetrantie-zie en Erika Check Hayden ‘ s artikel over ExAC. Je kunt mijn en Sonia ‘ s persoonlijke reis met deze studie hier voorlezen.,Multiple sclerose (MS) is een complexe ziekte met vele genetische risicofactoren , maar er is geen Mendeliaanse vorm van de ziekte bekend. Eerder dit jaar meldde een studie dat een missense variant in een nucleaire hormoonreceptor — NR1H3 R415Q-de allereerste Mendeliaanse vorm van MS veroorzaakt . Deze claim was gebaseerd op dominante segregatie met ziekte in twee families, maar de Lod score was slechts 2.2 — onder de drempel voor genoom-brede betekenis in familie linkage studies, die meer als 3.0 of 3.6 . En de variant in kwestie heeft een allelfrequentie van 0.,031% bij niet-Finse Europeanen. Dat klinkt misschien niet als een hoge allelfrequentie, maar het blijkt veel te hoog te zijn voor deze variant om Mendeliaanse MS te veroorzaken .
zijn van mening dat lidstaten een levenslange risico (in de algemene bevolking) hebben van 0,25% bij vrouwen en 0,14% bij mannen . Als 0,06% van de mensen in de algemene bevolking zijn r415q heterozygotes, en als zelfs de helft van hen ging op MS te ontwikkelen, dan is deze variant alleen zou goed zijn voor 0,03% van de bevolking ontwikkeling van MS.dus als een totaal van 0,25% van de mensen ontwikkelen MS, dan ongeveer 12% van hen moet deze variant hebben., In plaats daarvan werd de variant slechts gevonden bij 1 persoon uit een casusreeks van 2.053 MS-patiënten .
Dit werkt uit tot een allelfrequentie van 0,024% in gevallen, of 0,049% als we toestaan dat 2 gevallen worden geteld in de reeks gevallen. Dit is niet significant hoger dan de frequentie in ExAC. Maar als deze variant MS veroorzaakt, moet het vaker voorkomen in gevallen – veel vaker. Onthoud onze herschikte formule eerder: P (D/A)|P(D) = P(A/D) / P(A). Dit betekent dat als een variant het risico met X-voudig verhoogt, het X keer vaker voorkomt in controles. Dus als het basisrisico van MS 0 is.,25% en deze variant is 50% penetrant, het moet 50/zijn.25 = 200 maal vaker voor in gevallen dan controles. Als het zelfs 10% penetrantie had, zou het nog steeds 10/moeten zijn.25 = 40 keer vaker in gevallen dan in controles. Als alternatief kun je denken in termen van odds ratio ‘ s in plaats van waarschijnlijkheden. De 0,25% levenslange risico in de algemene bevolking betekent 1:399 odds, en als R415Q verleende 50% levenslange risico, dat zou 50:50 odds. (50/50)/(1/399) = 399, dus de odds ratio voor R415Q zou 399 moeten zijn om deze variant 50% penetrantie te hebben.,
in plaats daarvan, als we onze formule toepassen met behulp van de R-code van eerder, uitgaande van 0,25% baseline risico en het baseren van de berekening op 2 allelen op 2.053 gevallen, versus 21 allelen in 33.369 ExAC individuen, vinden we dat de bovengrens van de 95% BI op penetrantie 2,2% is. Dus zelfs als R415Q werden geassocieerd met MS-risico, kon het niet meer dan 2.2% levenslange risico van het ontwikkelen van MS .,
in hun formele respons en in PubMed Commons stelden de auteurs een vergelijking op met LRRK2 G2019S bij de ziekte van Parkinson, waarvan iedereen het eens is dat het pathogeen is, maar die ook in ExAC wordt aangetroffen en slechts een bescheiden odds ratio heeft, geschat op 9,6 . Voor die variant werkt de wiskunde. De ziekte van Parkinson komt minstens een orde van grootte vaker voor dan MS, met levenslange risico geschat ergens van 3,7% tot 6,7% . Deze orde van grootte grotere prevalentie betekent dat de ~ 10-voudige verrijking die is waargenomen-LRRK2 G2019S in ruwweg 0 wordt gevonden.,1% van de controles en 1% van de gevallen-is ruwweg consistent met het gerapporteerde ~32% levenslange risico van Parkinson toegekend door deze variant . Deze kwantitatieve details zijn van belang, en zijn verschillend voor elke variant en elke ziekte. Dat is de reden waarom de formules besproken in dit bericht nuttig zijn, ook al bieden ze alleen zeer ruwe schattingen en zijn onderworpen aan verschillende voorbehouden, zoals hieronder uitgelegd.
afwijkingen
In beide hierboven beschreven toepassingen werd informatie over de allelfrequentie gebruikt om een ruwe schatting van de penetrantie te krijgen., Bij prionziekte konden we aantonen dat varianten die eerder werden verondersteld zeer penetrant levensduur risico meer in de Orde van 0,1%, 1%, of 10%. In het NR1H3-verhaal was de allelfrequentie-informatie voldoende om aan te tonen dat de naar verluidt causale variant niet meer dan een paar procent levenslange risico kon geven.
maar proberen om allelfrequentie gegevens te gebruiken om een betere schatting van penetrantie te krijgen zou zeer uitdagend zijn. Zo zijn familiestudies het oneens over de penetrantie van PRNP E200K, met schattingen variërend van 60% tot 90% levenslange risico ‘ s ., Sinds de prion-studie uitkwam, heb ik een paar mensen uit e200k-families mij laten vragen of ExAC-gegevens kunnen helpen om te bepalen waar het risico binnen dit bereik ligt. Het antwoord is dat het helaas niet kan.
Hier zijn de belangrijkste redenen waarom ik denk dat alle penetrantieschattingen op basis van allelfrequentie moeten worden genomen met een korrel zout:
- als een variant zeer penetrant is, dan is het moeilijk om een casusreeks te verkrijgen die geen verwante individuen bevat. Als je case series related heeft, dan heb je technisch gezien geen onbevooroordeelde schatting van P(A|D).,
- als een ziekte fataal is, dan is het moeilijk om een populatie controle reeks te verkrijgen die niet op zijn minst enigszins uitgeput is van mensen met varianten die die ziekte veroorzaken. Dus dan heb je ook geen onbevooroordeelde schatting van P(A).
- vergelijkingen van gevallen-en controle-allelfrequentie zijn kwetsbaar voor verwarring door populatiestratificatie. In de prion-studie hadden we geen genoombrede SNP-gegevens over gevallen, dus was er geen manier om dit perfect te controleren.,veel causale varianten voor zeldzame ziekten zijn zo zeldzaam dat zelfs met ExAC, we nog niet voldoende nauwkeurige schattingen van de allelfrequentie hebben om beter dan een ruw antwoord te geven.
Dit gezegd zijnde, is de schatting van de allelfrequentie op basis van een populatie nog steeds een goede manier om een ruwe schatting van de penetrantie van de Orde van grootte te krijgen en om te controleren of een genetische variant aannemelijk Causaal kan zijn voor een zeldzame ziekte.