RNA-sequencing (RNA-seq) heeft een brede waaier van toepassingen, en er is geen optimale pijplijn voor alle gevallen. Wij herzien alle belangrijke stappen in RNA-seq gegevensanalyse, met inbegrip van kwaliteitscontrole, lezen uitlijning, kwantificering van gen en transcript niveaus, differentiële genuitdrukking, het functionele profileren, en geavanceerde analyse. Ze zullen later worden besproken.
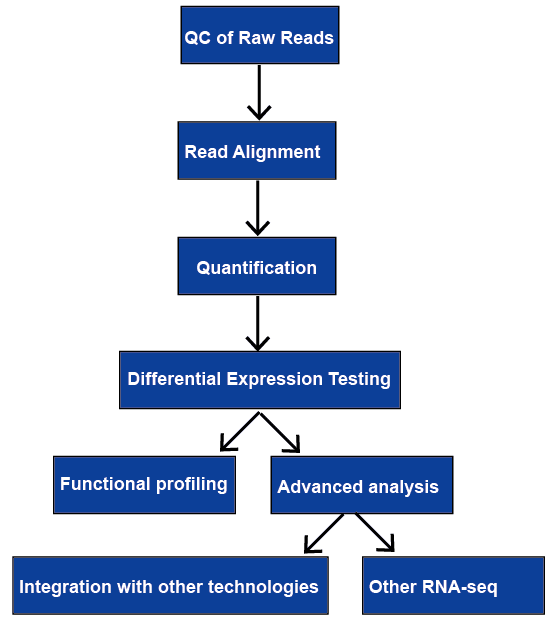
figuur 1. De algemene workflow van RNA-seq analyse.,
Quality control of raw reads
Quality control of RNA-seq raw reads bestaat uit analyse van sequentiekwaliteit, GC-inhoud, adapterinhoud, oververtegenwoordigde k-mers en gedupliceerde reads, gericht op het detecteren van sequentiefouten, contaminaties en PCR-artefacten. Lees kwaliteit daalt naar de 3 ‘ einde van Leest, basissen met lage kwaliteit, daarom moeten ze worden verwijderd om mappability te verbeteren., Naast de kwaliteit van ruwe gegevens omvat kwaliteitscontrole van ruwe leest ook de analyse van leesuitlijning (leesuniformiteit en GC-inhoud), kwantificering (3’ bias, biotypes en lage tellingen) en reproduceerbaarheid (correlatie, hoofdcomponentanalyse en batch-effecten).
Tabel 1. De hulpmiddelen voor kwaliteitscontrole van RNA-seq ruwe leest.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Ongeacht of een genoom of transcriptome verwijzing beschikbaar is, leest kan uniek in kaart brengen of aan veelvoudige posities in de verwijzing worden toegewezen, die als multi-in kaart gebrachte leest of multireads worden bedoeld. Genomic multireads zijn over het algemeen toe te schrijven aan herhaalde opeenvolgingen of gedeelde domeinen van paralogous genen. Transcriptome multi-mapping komt vaker voor als gevolg van genisovormen. Daarom, transcript identificatie en kwantificering zijn belangrijke uitdagingen voor alternatief uitgedrukt genen., Wanneer een verwijzing niet beschikbaar is, worden RNA-seq gelezen geassembleerd de novo gebruikend pakketten zoals SOAPdenovo-Trans, Oases, Trans-ABySS, of drie-eenheid. PE streng-specifieke en lange lengte leest de voorkeur omdat ze meer informatief zijn. De opkomende lang-gelezen technologieën, zoals het rangschikken van PacBio SMRT en Nanopore rangschikken, kunnen transcripten van volledige lengte voor de meeste genen produceren.
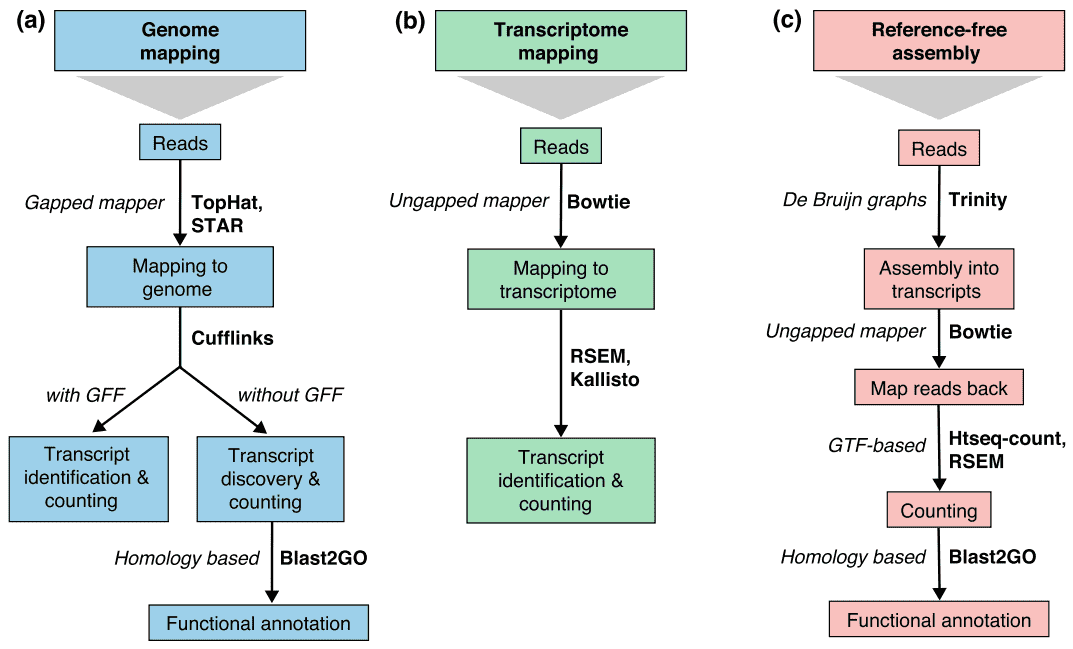
Figuur 2. Drie basisstrategieën voor RNA-seq gelezen mapping (Conesa et al. 2016)., Afkortingen: GFF, Algemeen Eigenschappenformaat; GTF, het formaat van de genoverdracht; RSEM,RNA-seq door Verwachtingsmaximalisatie.
Tabel 2. De vergelijking van genoom-gebaseerde en de novo assemblagestrategieën voor RNA-seq-analyse.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Manchetknopen | ontworpen om gebruik te maken van PE leest, en kan GTF-informatie gebruiken om uitgedrukte transcripten te identificeren, of kan transcripten de novo afleiden uit de mapping data alleen. |
| RSEM | Quantify expression from transcriptome mapping. alloceer multi-mapping reads tussen transcript en output binnen-sample genormaliseerde waarden gecorrigeerd voor het sequencen van vooroordelen., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
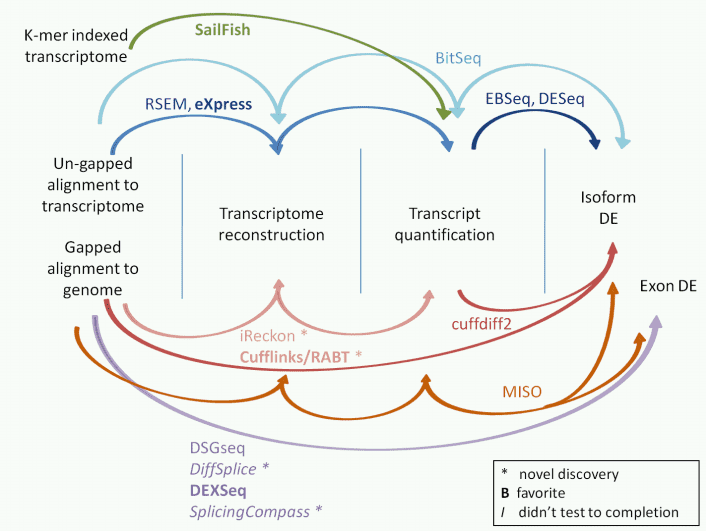
Figure 3. The tools for isoform expression quantification.,
differentiële expressietest
differentiële expressietest wordt gebruikt om te beoordelen of één gen in één toestand differentieel tot expressie wordt gebracht in vergelijking met de andere(s). Het normaliseren van methodes moeten worden goedgekeurd alvorens verschillende steekproeven te vergelijken. RPKM en TPM normaliseren weg de belangrijkste factor, sequencing diepte. TMM, DESeq en UpperQuartile kunnen zeer variabele en/of sterk uitgedrukte functies negeren., Andere factoren die interfereren met intra-sample vergelijkingen omvatten transcript lengte, positionele vooroordelen in de dekking, gemiddelde fragment grootte, en GC inhoud, die kan worden genormaliseerd door tools, zoals DESeq, edgeR, baySeq, en NOISeq. Batch effecten kunnen nog steeds aanwezig zijn na normalisatie, die kan worden geminimaliseerd door de juiste experimentele ontwerp, of verwijderd door methoden zoals COMBAT of ARSyN.
Tabel 5. De normalisatie tools voor differentiële expressie testen.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., De veelvoudige hulpmiddelen van de bioinformatica zijn ontwikkeld om vanaf experimentele gegevens te ontdekken. De vergelijking van deze opsporingshulpmiddelen gebruikend RNA-seq gegevens werd geleid door Ding in 2017, en de resultaten worden getoond in Lijst 7. Ze hebben aangetoond dat TopHat en zijn downstream tool, FineSplice, de snelste tools zijn, terwijl PASTA het traagste programma is. Bovendien kan AltEventFinder het hoogste aantal knooppunten detecteren, en RSR detecteert het laagste aantal knooppunten. Andere hulpmiddelen, zoals TopHat, zijn waarschijnlijk vals-positieve degenen te ontdekken., Van de twee tools die differentieel gesplitste isovormen detecteren, is rMATS sneller dan rSeqDiff, maar detecteert minder differentieel gesplitste isovormen dan rSeqDiff.
Tabel 7. Gedetecteerd als types of differentieel gesplitste isovormen van deze tools (Ding et al. 2017).,
visualisatie
Er zijn veel Bioinformatica-tools voor de visualisatie van RNA-seq-gegevens, waaronder genoombrowsers, zoals ReadXplorer, UCSC-browser, Integrative Genomics Viewer (IGV), Genoomkaarten, Savant, tools die speciaal zijn ontworpen voor RNA-seq-gegevens, zoals RNAseqViewer, evenals enkele pakketten voor differentiaalgenexpressieanalyse die de visualisatie mogelijk maken, zoals DESeq2 en DEXseq in Bioconductor. Pakketten, zoals CummeRbund en Sashimi percelen, zijn ook ontwikkeld voor visualisatie-exclusieve doeleinden.,
functionele profilering
De laatste stap in een standaard transcriptomica-studie is over het algemeen de karakterisering van de moleculaire functies of routes waarbij differentieel tot expressie gebrachte genen betrokken zijn. De ontologie van het gen, Bioconductor, DAVID, of Babelomics bevatten annotatiegegevens voor de meeste modelspecies, die voor functionele annotatie kunnen worden gebruikt. Zoals voor nieuwe afschriften, kunnen de eiwit-coderende afschriften functioneel worden geannoteerd gebruikend orthologie met behulp van gegevensbestanden zoals SwissProt, Pfam, en InterPro., De ontologie van het gen (GO) staat voor enige uitwisselbaarheid van functionele informatie tussen orthologs toe. Blast2GO is een populaire tool die massale annotatie van volledige transcriptome mogelijk maakt tegen een verscheidenheid aan databases en gecontroleerde woordenlijsten. Het rfam-gegevensbestand bevat de meeste goed-gekarakteriseerde families van RNA die voor functionele annotatie van lange niet-codeert RNAs kunnen worden gebruikt.
geavanceerde analyse
De geavanceerde analyse van RNA-seq omvat gewoonlijk andere RNA-seq en integratie met andere technologieën, die in Figuur 4 wordt geschetst., Meer informatie over toepassingen van RNA-seq, gelieve te bekijken Dit artikel toepassingen van RNA-Seq.
Figuur 3. De geavanceerde analyse van RNA-seq gegevens.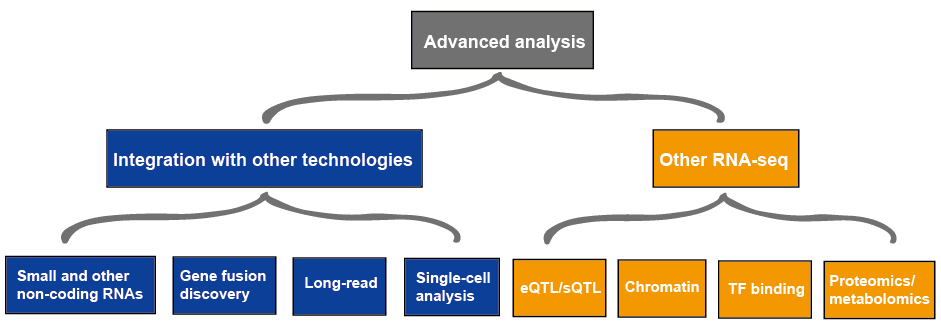
onze ervaren Bioinformatica wetenschappers zijn bedreven in het gebruik van de geavanceerde Bioinformatica tools om te gaan met de talrijke sequenties gegenereerd door de volgende en derde generatie sequencing. Wij bieden zowel het rangschikken als bioinformatics diensten voor genomics, transcriptomics, epigenomics, microbiële genomics, single-cell rangschikken, en PacBio SMRT rangschikken.