Il mese scorso sono stato invitato a parlare al Centers for Mendelian Genomics (CMG) Analysis and Methods Development meeting su “Stima basata sulla popolazione della penetranza nelle malattie rare”. Ecco la versione post sul blog del mio discorso.
che cos’è la penetranza e perché ci interessa?
La penetranza è la probabilità di sviluppare una particolare malattia data da un particolare genotipo., Si può parlare di penetranza dipendente dall’età, quindi la percentuale di persone con il genotipo che sviluppano la malattia entro i 40 anni, entro i 50 anni e così via; Di solito parlo in termini di rischio per tutta la vita, ovvero la probabilità che tu sviluppi la malattia prima di morire. Inerente a questo è che, per le malattie ad esordio adulto, il rischio di vita non può mai essere del 100%, perché potresti sempre morire prima di qualcos’altro.
La penetranza è estremamente importante per gli individui sottoposti a test genetici predittivi — la prima domanda di molte persone è: “questo significa che avrò sicuramente la malattia?”., Eppure è spesso molto difficile da venire da una stima ferma di penetranza.
metodi tradizionali per stimare la penetranza
In un mondo ideale, il modo giusto per stimare la penetranza sarebbe quello di accertare, dalla nascita, una grande coorte di persone con un particolare genotipo, seguirli fino a quando tutti sono morti di qualcosa o altro, e poi chiedere quanti hanno mai sviluppato la malattia prima di morire. Dal momento che la tecnologia di genotipizzazione è stata inventata meno di una vita umana fa, questo non è mai stato fatto per nessuna malattia.
Invece, i ricercatori usano spesso metodi basati sulla famiglia per stimare la penetranza., Uno studio tipico guarderebbe a tutti coloro che sono stati osservati con il genotipo dato, e chiedere quanti hanno diease, o quanti hanno la malattia da una certa età. I metodi basati sulla famiglia soffrono di pregiudizi pervasivi di accertamento ., accertato, sulla base di presentare con malattia
Come un esempio di questo ultimo punto, la genetica delle malattie da prioni, solo il 23% di persone a rischio di scegliere test genetici predittivi , e nel pedigree dati che ho avuto accesso, sapevamo che i genotipi di solo il 22% dei soggetti a rischio .,
Tutti i fattori sopra elencati lavorano nella stessa direzione, tendendo a gonfiare la propria stima di penetranza.
I ricercatori sono a conoscenza di questi problemi da molto tempo e hanno proposto alcune soluzioni. Come esempio, il metodo kin-coorte comporta l’accertamento di individui sani in modo casuale da una popolazione, genotipizzazione loro, prendendo una storia familiare, e confrontando le curve di sopravvivenza dei loro parenti di primo grado., Questa è una soluzione molto intelligente, ma si basa sulla possibilità di accertare un numero sufficiente di persone con un genotipo che causa la malattia senza accertare la presenza di malattia. Quindi ha funzionato per le varianti BRCA1 e BRCA2 negli ebrei Ashkenazi americani, ma per molte condizioni genetiche più rare, è poco pratico, perché avresti bisogno di reclutare decine o centinaia di migliaia di persone per trovare anche un individuo con un genotipo di interesse.,
metodi basati sulla popolazione
Per tutti i motivi sopra descritti, è molto utile avere metodi ortogonali basati sulla popolazione, per porre domande sulla penetranza. La prima intuizione chiave qui è che una variante genetica completamente penetrante non dovrebbe essere più comune nella popolazione rispetto alla malattia che provoca. Applicare questa logica in pratica significa che hai bisogno di buone stime della frequenza degli alleli anche per varianti non comuni, e questo è stato difficile da trovare fino a poco tempo fa. ExAC, un database di variazione genetica in 60.706 esomi umani, offre nuove opportunità ., Molti individui in ExAC sono stati accertati come casi o controlli per varie malattie comuni e complesse, ma nessuno è stato accertato per la malattia mendeliana, quindi ExAC è un buon database di riferimento per lo studio della maggior parte delle malattie genetiche.
Fornendo informazioni sulla frequenza degli alleli nella popolazione generale, ExAC, come i precedenti database di riferimento come ESP , ha chiarito che la genetica clinica ha un grosso problema: molte varianti segnalate per causare malattie genetiche in realtà non causano malattie genetiche, o almeno non la maggior parte del tempo.,
Due database — HGMD e ClinVar — raccolgono affermazioni dalla letteratura e dai laboratori clinici che affermano che una particolare variante genetica causa una particolare malattia genetica. All’ultimo conteggio, c’erano oltre 100.000 varianti genetiche che causano malattie uniche in questi database. La persona media in ExAC ha 54 di loro . Ovviamente, la persona media in realtà non ha 54 malattie genetiche., Naturalmente, gran parte di questo eccesso è causato da un piccolo numero di varianti selvaggiamente ad alta frequenza che ovviamente non causano alcuna malattia genetica, e gran parte di esso può essere riferito varianti recessive trovate in uno stato eterozigote in ExAC. Ma anche se guardiamo solo alle varianti nei geni delle malattie dominanti con una frequenza allele di <1%, vediamo ancora 0,89 varianti patogene per persona, e chiaramente non è il caso che ~90% delle persone abbia una malattia genetica dominante., Quindi, attraverso lo spettro di frequenza degli alleli, ci sono molte varianti patogene che non sono così patogene. Quando Anne O’Donnell e io abbiamo esaminato le varianti patogene con le più alte frequenze di allele in ExAC, e abbiamo chiesto come fossero riusciti a essere classificati erroneamente come patogeni, abbiamo scoperto che il più delle volte il problema risaliva a un articolo in letteratura che aveva fatto una richiesta di patogenicità basata su prove insufficienti.
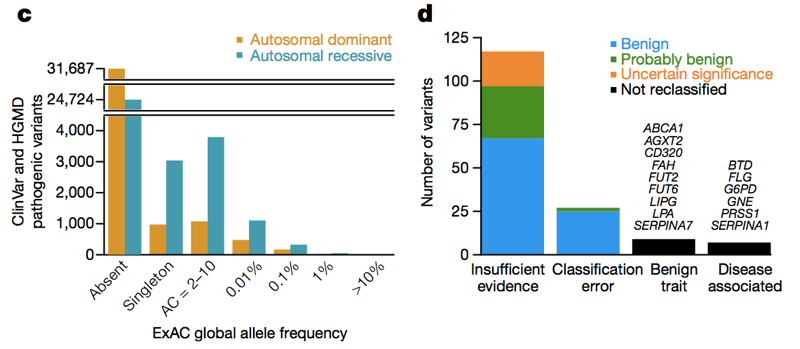
Sopra: Figure 3C e 3D da ., Attraverso lo spettro di frequenza dell’allele e sia nei geni dominanti che recessivi della malattia, ci sono molte varianti patogene riferite che compaiono in ExAC. Di alta (>1%) frequenza riferito varianti patogene, alcuni sono genuinamente patogeni, alcuni sono genuinamente associati a tratti ma il tratto è benigno, e alcuni sono errori di annotazione nei database-ma la maggior parte si basa su letteratura con prove insufficienti.,
Le informazioni sulla frequenza allele di ExAC hanno ora permesso di riclassificare oltre 200 varianti genetiche da patogene a benigne, probabilmente benigne o di significato incerto . Questi tipi di riclassificazioni a volte innescano il pushback dagli autori originali che hanno proposto che una variante causi una malattia genetica, che potrebbe sostenere che una variante potrebbe ancora essere patogena, ma con penetranza incompleta. Ma quanto può essere” incompleta ” la penetranza incompleta?, Dobbiamo ottenere quantitativi, perché se il rischio di vita è al massimo dell ‘ 1%, allora è ancora ragionevole dire che una variante “causa” una malattia genetica o è “patogena”? Mentre le informazioni sulla frequenza degli alleli non possono mai dimostrare che una variante non ha alcuna associazione con la malattia, può porre limiti su quale potrebbe essere la possibile penetranza, e in molti casi, anche per varianti abbastanza rare, è possibile dimostrare che non c’è modo che una variante conferisca un livello di rischio lontanamente vicino al 100%.,
Per ottenere quantitativi, dobbiamo estendere la nostra precedente osservazione — che una variante genetica completamente penetrante non dovrebbe essere più comune nella popolazione rispetto alla malattia che causa. Questo è tutto semplice matematica e genetica delle popolazioni, ma troppo spesso non viene applicato nella pratica. Qui ci sono due modi in cui possiamo pensare alla frequenza degli allele quando facciamo inferenze sulla patogenicità e penetranza.
massima frequenza allele credibile
Dire che si sta studiando l’esoma di un paziente con malattia mendeliana e cercando di identificare la variante causale., Il mio collega James Ware ha messo a punto una strategia per filtrare che exome contro le informazioni di frequenza allele in ExAC, sfruttando la seguente logica., La massima frequenza allele che è plausibile per una variante a causa di una malattia genetica dominante è pari alla prevalenza della malattia volte l’eterogeneità allelica (la proporzione di casi attribuibili a una variante) diviso per penetranza (meno penetranti le varianti possono essere più comune), diviso per 2 (perché ci sono diploidi):
\
Per esempio, malattia da prioni cause in 1 in 5.000 morti, e la variante più comune (E200K) si trova nel 5% dei casi , quindi al 100% penetranti variante non può avere allele frequenza maggiore di 0,0005% (1 in 200.000) ., La cardiomiopatia colpisce 1 persona su 500, la variante più comune si trova in <2% dei casi, quindi una variante penetrante al 50% non può avere una frequenza allele superiore allo 0,004% . La formula per le malattie recessive è una tacca più complicato, ma James ha anche lavorato fuori ed è descritto in .
Quindi , mentre storicamente le persone hanno spesso filtrato varianti con una frequenza allele>0.1% quando si cerca di identificare la causa di una malattia dominante, possiamo effettivamente essere molto più rigorosi., L’avvertenza è che a bassi conteggi di allele, la nostra capacità di stimare la frequenza dell’allele è limitata dalla varianza di campionamento. Ad esempio, se guardiamo alle varianti osservate con una frequenza allele dell ‘1% tra gli europei in ESP, queste varianti hanno anche una frequenza dell’ 1% tra gli europei ExAC. Ma le varianti con una frequenza dello 0,1% in ESP tendono ad essere leggermente più rare in ExAC, e la maggior parte dei singleton (varianti viste esattamente una volta in ESP) non riappaiono una seconda volta in ExAC.
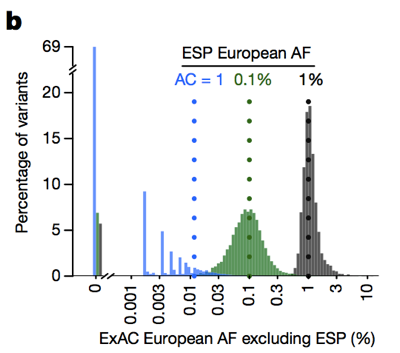
Sopra: Figura 3B da . Più basso è il conteggio allele, meno buona una stima della frequenza allele fornisce.,
Pertanto, più basso è il conteggio degli alleli, più conservativo dobbiamo essere. Abbiamo ideato un framework per farlo usando il limite superiore del 95% della distribuzione di Poisson su quanti alleli potrebbero essere osservati a una data frequenza e hanno valori pre-calcolati per tutti ExAC (disponibili su FTP) che è possibile utilizzare — leggi di più sui metodi in . James ha anche creato questa applicazione web a portata di mano che permette di esplorare ciò che la “massima frequenza allele credibile” dovrebbe essere per la vostra malattia di interesse.,
Inerente a questo approccio è che minore è la penetranza di una variante, maggiore è la frequenza che potrebbe avere nella popolazione generale. Ma devi anche capire che se la penetranza è piuttosto bassa, diciamo, inferiore al 10%, allora anche l’utilità clinica di quella variante è bassa. James e Nicky Whiffin hanno presentato dati per dimostrare che quasi tutta l’utilità clinica del sequenziamento in cardiomiopatia proviene da varianti con una frequenza di <0,001% — le varianti più comuni contribuiscono cumulativamente a un rischio minimo, se presente .,
stima e limiti del rischio di vita
Ricorda che la penetranza è la probabilità di malattia data da un particolare genotipo. Oppure, se consideriamo un modello allelico piuttosto che genotipico, la probabilità di malattia dato un particolare allele. Possiamo scrivere questo come P (D|A). Una volta fatto, diventa chiaro che, per teorema di Bayes,
\
Ognuno di questi termini ha un significato particolare:
Si noti qui che “controlli di popolazione” significa un gruppo non selezionato per la presenza, né per l’assenza, della malattia. Solo una fetta della popolazione generale.
Quindi:
\
Questa logica non è una novità., L’uso del teorema di Bayes per stimare il rischio di malattia risale almeno alla stima del rischio di cancro nei fumatori e la sua applicazione alla genetica è stata considerata per quasi altrettanto tempo . Ma perché questa equazione funzioni per le malattie rare, hai bisogno di stime abbastanza buone della frequenza degli allele di controllo dei casi e della popolazione, e quelle sono state difficili da trovare fino a poco tempo fa. Quindi, grazie a ExAC, ci sono un numero crescente di situazioni in cui questa equazione è rilevante.
Ecco il codice R che ho scritto (originariamente qui) per stimare la penetranza in base a questa formula.,
Se non si desidera eseguire il codice R da soli, James Ware ha implementato nella scheda “penetrance” di questa applicazione web in modo da poter semplicemente collegare i numeri nel browser.
Per stimare gli intervalli di confidenza del 95% sulla penetranza, ho adottato l’approccio di . Si immette il conteggio degli alleli (AC) e il numero di individui (N) per i casi e i controlli e il limite superiore dell’IC al 95% viene calcolato in base all’IC al 95% superiore della distribuzione binomiale per la frequenza dell’allele del caso e l’IC al 95% inferiore per i controlli., Viceversa, il limite inferiore della penetranza si basa sul limite inferiore della frequenza dell’allele di caso e sul limite superiore della frequenza dell’allele di controllo. Si potrebbe giustamente cavillare che, poiché questa formula utilizza CIs al 95% su entrambi i valori di frequenza allele, gli intervalli di confidenza risultanti sono più grandi di quanto dovrebbero essere. Si potrebbe anche giustamente cavillare che la distribuzione binomiale non è un buon stimatore a bassi conteggi di allele, a causa del pregiudizio illustrato nella Figura 3B mostrata sopra (e certamente non applicherei mai questa formula a singleton — varianti osservate solo una volta in ExAC)., Ma alla fine della giornata, per motivi che discuterò più vicino alla fine di questo post, questa formula è davvero meglio utilizzata per ottenere un campo da baseball, stima dell’ordine di grandezza della penetranza. Se stai cercando una stima puntuale estremamente precisa della penetranza, questo intero approccio probabilmente non funzionerà comunque per te.
Se si riorganizza l’equazione, un altro modo di pensarci è:
\
Ciò significa che l’aumento del rischio tra le persone con un genotipo è proporzionale al rapporto tra caso e frequenza allele di controllo della popolazione., Quindi una variante che aumenta il rischio di 200 volte dovrebbe essere 200 volte più comune tra i casi di quanto non sia nella popolazione generale. (Si noti che questo rapporto di frequenze allele è leggermente diverso da odds ratio anche se le due misure convergono per varianti molto rare.)
applicazione alla malattia da prioni
Abbiamo camminato attraverso questa logica in uno studio che abbiamo pubblicato all’inizio di quest’anno, quantificando la penetranza delle varianti della malattia da prioni ., Mi interessa la malattia da prioni per una ragione personale-mia moglie ospita una variante patogena in PRNP-ma si scopre che la malattia da prioni è anche un ottimo banco di prova per usare la logica di cui sopra per stimare la penetranza. Nessuno degli individui in ExAC v1 è stato accertato sulla malattia neurodegenerativa, quindi ExAC è davvero un buon set di dati di controllo della popolazione per la malattia da prioni. E poiché le malattie da prioni sono “notificabili”, i centri di sorveglianza nazionali hanno un accertamento dei casi eccezionalmente buono, e grazie alla loro generosità nella condivisione dei dati, siamo stati in grado di accumulare un set di dati di 10.460 casi sequenziati.,
Abbiamo scoperto che le>60 varianti segnalate per causare la malattia da prioni hanno cumulativamente 52 alleli in ExAC. Ciò significa che quasi 1 persona su 1.000 ha una di queste varianti, e quindi queste varianti sono cumulativamente molto più comuni di tutte le malattie da prioni (che causano ~1 ogni 5.000 morti), per non parlare di tutte le malattie genetiche da prioni (solo ~15% dei casi sono genetiche). Questo è sufficiente per dirci che non tutte queste varianti possono essere completamente penetranti. Al fine di determinare quali varianti erano i colpevoli, abbiamo confrontato con la serie di casi., Varianti con eccellente evidenza precedente di patogenicità (segregazione mendeliana e modelli murini) erano comuni nei casi e assenti da ExAC, coerenti con penetranza completa o quasi completa. La maggior parte del numero di alleli in eccesso in ExAC è stato contribuito da varianti che erano non comuni nei casi e avevano una debole evidenza precedente di patogenicità — queste varianti sono probabilmente benigne o contribuiscono solo a un basso rischio. Almeno tre varianti apparivano intermedie, poiché erano troppo comuni nei controlli per la piena penetranza, ma ancora arricchite nei casi sopra i controlli.,
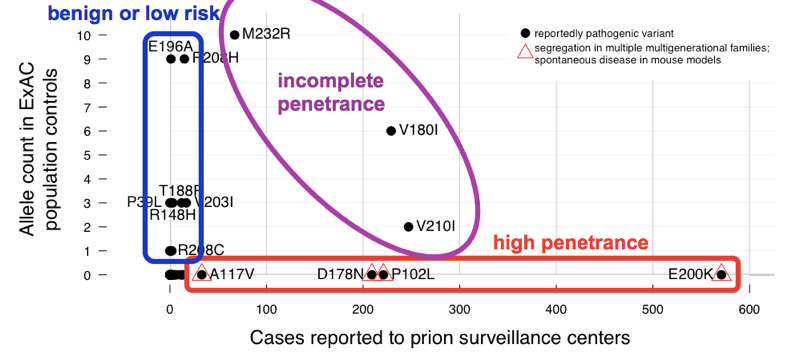
Sopra: una versione annotata di Figura 2 da .
Quando abbiamo stimato la penetranza per ogni variante, usando la formula P(D|A) sopra, abbiamo scoperto che esiste un intero spettro di penetranza per le varianti PRNP.
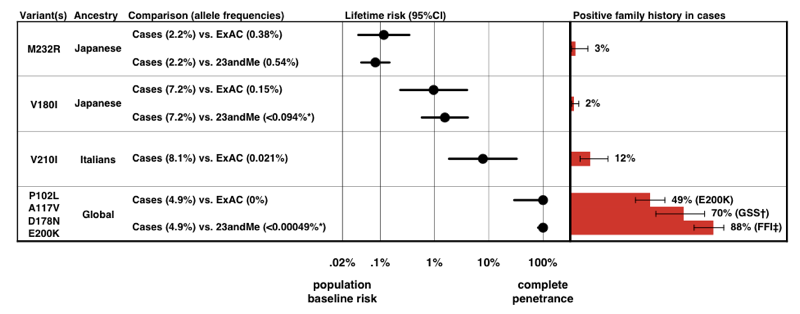
Sopra: Figura 3 da .
Nota la scala sull’asse x — per una malattia così rara che la probabilità precedente di svilupparla è solo dello 0,02%, anche un aumento di 50 volte del rischio è solo dell ‘ 1% di rischio per tutta la vita., Rassicurante, la penetranza stima che deriviamo da informazioni di frequenza allele da solo accordo abbastanza bene con la percentuale di casi che presentano con una storia familiare positiva.
Questo lavoro ha già portato a un cambiamento nella prognosi per alcuni individui che erano stati originariamente consigliati che erano a rischio di varianti ad alta penetranza-vedi ed Erika Controlla l’articolo di Hayden su ExAC. Puoi leggere il mio e il viaggio personale di Sonia con questo studio qui.,
applicazione a NR1H3
La sclerosi multipla (SM) è una malattia complessa con molti fattori di rischio genetici , ma non è nota alcuna forma mendeliana della malattia. All’inizio di quest’anno, uno studio ha riferito che una variante missense in un recettore ormonale nucleare — NR1H3 R415Q — causa la prima forma mendeliana di SM . Questa affermazione era basata sulla segregazione dominante con malattia in due famiglie, ma il punteggio LOD era solo 2.2 — sotto la soglia per il significato a livello del genoma negli studi di legame familiare, che è più simile a 3.0 o 3.6 . E la variante in questione ha una frequenza allele di 0.,031% negli europei non finlandesi ExAC. Potrebbe non sembrare una frequenza allele alta, ma risulta essere troppo alta perché questa variante causi la MS mendeliana .
Si consideri che la SM ha un rischio per tutta la vita (nella popolazione generale) dello 0,25% nelle donne e dello 0,14% negli uomini . Se lo 0,06% delle persone nella popolazione generale sono eterozigoti R415Q, e se anche la metà di loro ha continuato a sviluppare la SM, allora questa variante da sola rappresenterebbe lo 0,03% della popolazione che sviluppa la SM.Quindi se un totale di 0,25% delle persone sviluppa la SM, allora circa il 12% di loro dovrebbe avere questa variante., Invece, la variante è stata trovata solo in 1 individuo su una serie di casi di 2.053 pazienti con SM .
Questo funziona con una frequenza allele di 0,024% nei casi, o 0,049% se permettiamo di contare 2 casi nella serie di casi. Questo non è significativamente superiore alla frequenza in ExAC. Ma se questa variante causa la SM, dovrebbe essere più comune nei casi-molto più comune. Ricorda la nostra formula riorganizzata in precedenza: P(D|A)/P(D) = P(A|D)/P (A). Ciò significa che se una variante aumenta il rischio di X-fold, dovrebbe essere X volte più comune nei controlli. Quindi, se il rischio di base della SM è 0.,25% e questa variante è 50% penetrante, dovrebbe essere 50/.25 = 200 volte più comune nei casi rispetto ai controlli. Se avesse anche penetranza 10%, dovrebbe essere ancora 10/.25 = 40 volte più comune nei casi che nei controlli. In alternativa, si può pensare in termini di odds ratio invece di probabilità. Il rischio di vita 0.25% nella popolazione generale significa 1: 399 probabilità, e se R415Q conferito 50% rischio di vita, che sarebbe 50: 50 probabilità. (50/50)/(1/399) = 399, quindi il rapporto di probabilità per R415Q dovrebbe essere 399 in modo che questa variante abbia penetranza del 50%.,
Invece, se applichiamo la nostra formula usando il codice R di prima, assumendo il rischio di base dello 0,25% e basando il calcolo su 2 alleli su 2.053 casi, rispetto a 21 alleli in 33.369 individui ExAC, troviamo che il limite superiore dell’IC al 95% sulla penetranza è del 2,2%. Quindi, anche se R415Q fosse associato al rischio di SM, non potrebbe conferire più del 2,2% di rischio per tutta la vita di sviluppare la SM .,
Nella loro risposta formale e in PubMed Commons gli autori hanno sollevato un confronto con LRRK2 G2019S nella malattia di Parkinson, che tutti sono d’accordo è patogeno, ma che si trova anche in ExAC e ha solo un modesto odds ratio, stimato a 9.6 . Per quella variante, la matematica funziona. La malattia di Parkinson è almeno un ordine di grandezza più prevalente della SM, con un rischio di vita stimato dal 3,7% al 6,7% . Questo ordine di grandezza maggiore prevalenza significa che l’arricchimento ~ 10 volte che è stato osservato — LRRK2 G2019S si trova in circa 0.,1% dei controlli e 1% dei casi-è approssimativamente coerente con il rischio di vita riportato ~32% di Parkinson conferito da questa variante . Questi dettagli quantitativi contano e sono diversi per ogni variante e ogni malattia. Ecco perché le formule discusse in questo post sono utili, anche se forniscono solo stime molto approssimative e sono soggette a diversi avvertimenti, come spiegato di seguito.
avvertenze
In entrambe le applicazioni sopra descritte, le informazioni sulla frequenza allele sono state utilizzate per ottenere una stima approssimativa della penetranza., Nella malattia da prioni, siamo stati in grado di dimostrare che le varianti precedentemente presunte altamente penetranti conferivano un rischio di vita più dell’ordine dello 0,1%, 1% o 10%. Nella storia NR1H3, le informazioni sulla frequenza degli alleli erano sufficienti per dimostrare che la variante causale riferito non poteva conferire più di un rischio di vita di pochi per cento.
Ma cercare di utilizzare i dati di frequenza allele per ottenere una stima più stretta della penetranza sarebbe molto impegnativo. Ad esempio, gli studi basati sulla famiglia non sono stati d’accordo sulla penetranza del PRNP E200K, con stime che vanno dal 60% al 90% del rischio di vita ., Da quando è uscito lo studio sui prioni, alcune persone delle famiglie E200K mi hanno chiesto se i dati ExAC possono aiutare a restringere dove il rischio è all’interno di questo intervallo. La risposta è, sfortunatamente, non può.
Ecco i motivi più importanti per cui penso che tutte le stime di penetranza basate sulla frequenza degli alleli debbano essere prese con un pizzico di sale:
- Se una variante è altamente penetrante, allora è difficile ottenere una serie di casi che non contenga individui correlati. Se la tua serie di casi ha relazioni, tecnicamente non hai una stima imparziale di P (A|D).,
- Se una malattia è fatale, allora è difficile ottenere una serie di controllo della popolazione che non sia almeno un po ‘ esaurita di persone con varianti che causano quella malattia. Quindi non hai nemmeno una stima imparziale di P(A).
- I confronti della frequenza degli alleli di caso e controllo sono vulnerabili alla confusione dovuta alla stratificazione della popolazione. Nello studio sui prioni, non avevamo dati SNP a livello genomico sui casi, quindi non c’era modo di controllare perfettamente per questo.,
- Molte varianti causali per le malattie rare sono così rare che anche con ExAC, non abbiamo ancora stime sufficientemente precise della frequenza degli alleli per dare una risposta migliore di una risposta approssimativa.
Detto questo, la stima della frequenza degli alleli basata sulla popolazione è ancora un buon modo per ottenere stime approssimative dell’ordine di grandezza della penetranza e per eseguire controlli di sanità mentale sul fatto che una variante genetica possa plausibilmente essere causale per una malattia rara.