RNA-sequencing (RNA-seq) ha una vasta gamma di applicazioni, e non esiste una pipeline ottimale per tutti i casi. Esaminiamo tutti i passaggi principali nell’analisi dei dati RNA-seq, incluso il controllo di qualità, l’allineamento di lettura, la quantificazione dei livelli di gene e trascrizione, l’espressione genica differenziale, la profilazione funzionale e l’analisi avanzata. Saranno discussi in seguito.
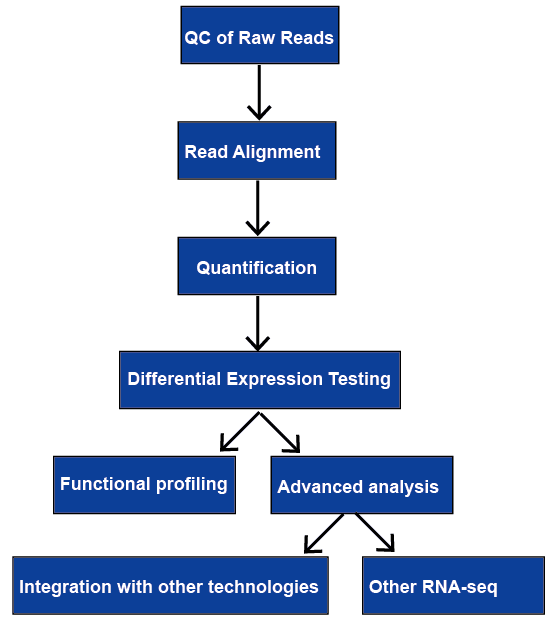
Figura 1. Il flusso di lavoro generale di analisi RNA-seq.,
Controllo di qualità delle letture raw
Il controllo di qualità delle letture raw RNA-seq consiste nell’analisi della qualità della sequenza, del contenuto GC, del contenuto dell’adattatore, dei k-mer sovrarappresentati e delle letture duplicate, dedicato alla rilevazione di errori di sequenziamento, contaminazioni e artefatti PCR. La qualità di lettura diminuisce verso l’estremità 3’ delle letture, basi con bassa qualità, pertanto, dovrebbero essere rimosse per migliorare la mappabilità., Oltre alla qualità dei dati grezzi, il controllo di qualità delle letture grezze include anche l’analisi dell’allineamento di lettura (uniformità di lettura e contenuto GC), della quantificazione (bias 3’, biotipi e conteggi bassi) e della riproducibilità (correlazione, analisi dei componenti principali ed effetti batch).
Tabella 1. Gli strumenti per il controllo di qualità di RNA-seq raw legge.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Indipendentemente dal fatto che sia disponibile un riferimento al genoma o al trascrittoma, le letture possono mappare in modo univoco o essere assegnate a più posizioni nel riferimento, che sono indicate come letture multi-mappate o multireads. I multiread genomici sono generalmente dovuti a sequenze ripetitive o domini condivisi di geni paralogici. La multi-mappatura del trascrittoma si presenta più spesso a causa delle isoforme geniche. Pertanto, l’identificazione e la quantificazione della trascrizione sono sfide importanti per i geni espressi in alternativa., Quando un riferimento non è disponibile, le letture RNA-seq vengono assemblate de novo utilizzando pacchetti come SOAPdenovo-Trans, Oasi, Trans-ABySS o Trinity. PE strand-specifica e di lunga lunghezza legge sono preferiti in quanto sono più informativo. Le tecnologie emergenti a lunga lettura, come il sequenziamento PacBio SMRT e il sequenziamento nanoporo, possono generare trascrizioni a lunghezza intera per la maggior parte dei geni.
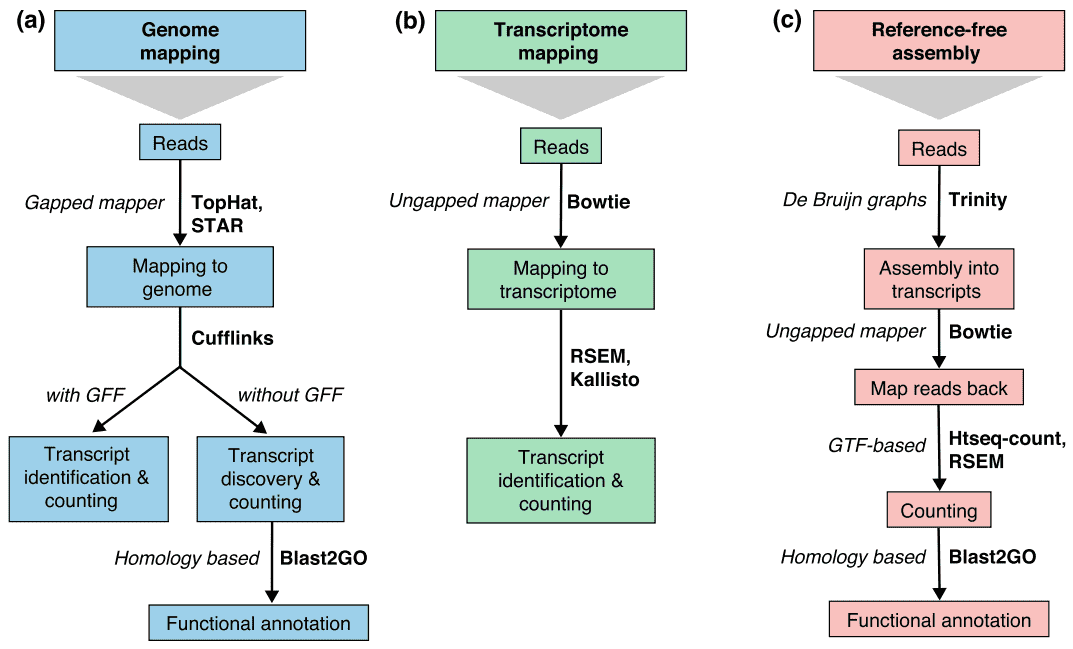
Figura 2. Tre strategie di base per la mappatura di lettura RNA-seq (Conesa et al. 2016)., Abbreviazioni: GFF, General Feature Format; GTF, gene transfer format; RSEM, RNA-seq per Massimizzazione delle aspettative.
Tabella 2. Il confronto delle strategie di assemblaggio genome-based e de novo per l’analisi RNA-seq.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Gemelli | Progettato per sfruttare PE legge, e può utilizzare le informazioni GTF per identificare trascrizioni espresse, o può dedurre trascrizioni de novo dai soli dati di mappatura. |
| RSEM | Quantifica l’espressione dalla mappatura del trascrittoma. Allocare letture multi-mapping tra trascrizione e output all’interno del campione valori normalizzati corretti per sequenziamento pregiudizi., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
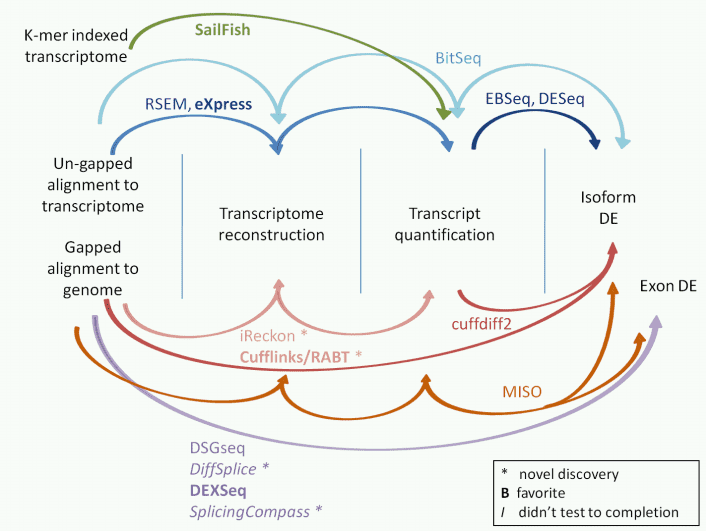
Figure 3. The tools for isoform expression quantification.,
Test di espressione differenziale
Il test di espressione differenziale viene utilizzato per valutare se un gene è espresso in modo differenziale in una condizione rispetto agli altri. I metodi di normalizzazione devono essere adottati prima di confrontare diversi campioni. RPKM e TPM normalizzare via il fattore più importante, sequenziamento profondità. TMM, DESeq e UpperQuartile possono ignorare caratteristiche altamente variabili e/o altamente espresse., Altri fattori che interferiscono con i confronti intra-campione riguardano la lunghezza della trascrizione, i pregiudizi posizionali nella copertura, la dimensione media del frammento e il contenuto GC, che possono essere normalizzati da strumenti come DESeq, edgeR, baySeq e NOISeq. Effetti batch possono essere ancora presenti dopo la normalizzazione, che può essere minimizzato da un adeguato disegno sperimentale, o rimossi con metodi come il combattimento o ARSyN.
Tabella 5. Gli strumenti di normalizzazione per il test di espressione differenziale.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Più strumenti bioinformatici sono stati sviluppati per rilevare COME da dati sperimentali. Il confronto di questi strumenti di rilevamento utilizzando i dati RNA-seq è stato condotto da Ding nel 2017 e i risultati sono mostrati nella Tabella 7. Hanno dimostrato che TopHat e il suo strumento a valle, FineSplice, sono gli strumenti più veloci, mentre la PASTA è il programma più lento. Inoltre, AltEventFinder può rilevare il maggior numero di giunzioni e RSR rileva il minor numero di giunzioni. Altri strumenti, come TopHat, sono suscettibili di rilevare quelli falsi positivi., Dei due strumenti che rilevano isoforme con giunzioni differenziate, rMATS è più veloce di rSeqDiff ma rileva meno isoforme con giunzioni differenziate rispetto a rSeqDiff.
Tabella 7. Rilevato come tipi o isoforme differenzialmente impiombate di questi strumenti (Ding et al. 2017).,
Visualizzazione
Ci sono molti strumenti bioinformatici per la visualizzazione di dati di RNA-seq, tra cui genome browser, come ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Genoma Mappe, Savant, strumenti specificamente progettati per i dati di RNA-seq, come RNAseqViewer, così come alcuni pacchetti di espressione genica differenziale di analisi che consentono la visualizzazione, ad esempio DESeq2 e DEXseq in Bioconductor. Pacchetti, come CummeRbund e trame Sashimi, sono stati sviluppati anche per scopi esclusivi di visualizzazione.,
Profilazione funzionale
L’ultimo passo in uno studio di trascrittomica standard è generalmente la caratterizzazione delle funzioni molecolari o dei percorsi in cui sono coinvolti geni espressi in modo differenziale. Gene Ontology, Bioconductor, DAVID o Babelomics contengono dati di annotazione per la maggior parte delle specie modello, che possono essere utilizzati per l’annotazione funzionale. Per quanto riguarda le nuove trascrizioni, le trascrizioni codificanti proteine possono essere annotate funzionalmente utilizzando l’ortologia con l’aiuto di database come SwissProt, Pfam e InterPro., L’ontologia genica (GO) consente una certa scambiabilità di informazioni funzionali tra gli ortologi. Blast2GO è uno strumento popolare che permette massiccia annotazione di trascrittoma completo contro una varietà di database e vocabolari controllati. Il database Rfam contiene la maggior parte delle famiglie di RNA ben caratterizzate che possono essere utilizzate per l’annotazione funzionale di RNA lunghi non codificanti.
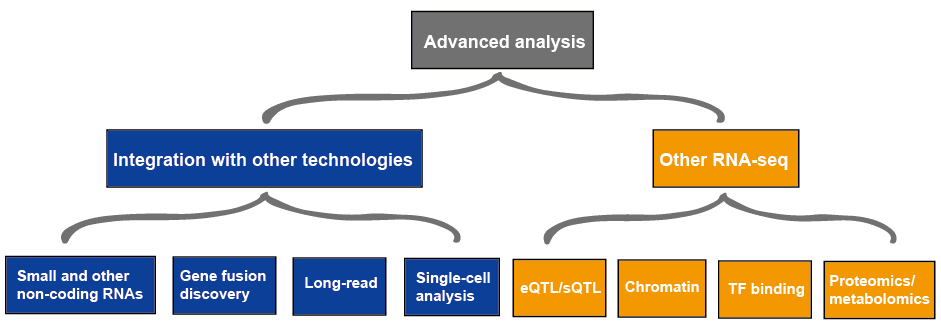
Analisi avanzata
L’analisi avanzata di RNA-seq di solito include altri RNA-seq e l’integrazione con altre tecnologie, che è delineata nella Figura 4., Maggiori informazioni sulle applicazioni di RNA-seq, si prega di consultare questo articolo Applicazioni di RNA-Seq.
Figura 3. L’analisi avanzata dei dati RNA-seq.
I nostri esperti scienziati di bioinformatica sono esperti nell’utilizzare gli strumenti bioinformatici avanzati per affrontare le numerose sequenze generate dal sequenziamento di prossima e terza generazione. Forniamo sia sequenziamento e bioinformatica servizi per la genomica, trascrittomica, epigenomica, genomica microbica, sequenziamento unicellulare, e PACBIO SMRT sequenziamento.