RNS-szekvenálás (RNS-seq) bioinformatikai munkafolyamata széles körű alkalmazásokkal rendelkezik, minden esetben nincs optimális csővezeték. Áttekintjük a főbb lépéseket RNS-seq adatok elemzését, beleértve a minőség-ellenőrzés, olvassa el igazítás, számszerűsítését gén transcript szinten, differenciál génexpresszió, funkcionális beidegződés speciális elemzés. Később megvitatják őket.
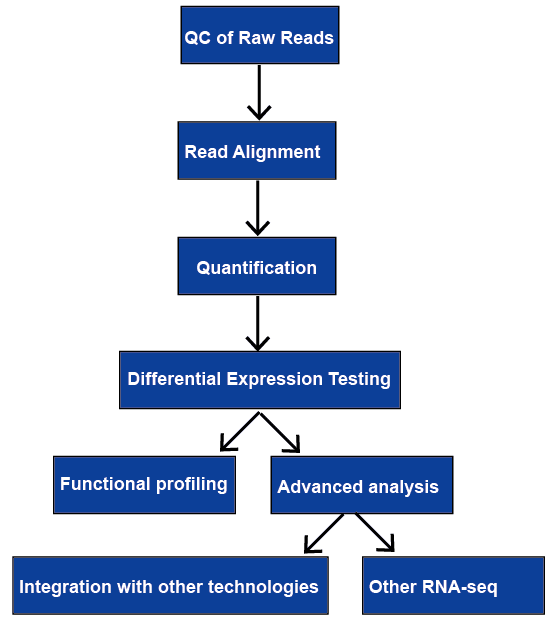
1.ábra. Az RNS-seq analízis általános munkafolyamata.,
minőségellenőrzés raw reads
minőségellenőrzés RNS-seq raw reads áll elemzése szekvencia minőség, GC tartalom, adaptertartalom, felülreprezentált k-mers, és duplikált olvasás, szentelt kimutatására szekvenálás hibák, szennyeződések, és PCR leletek. Az olvasási minőség csökken az olvasás 3 ” vége felé, alacsony minőségű alapokkal, ezért azokat el kell távolítani a leképezhetőség javítása érdekében., A nyers adatok minősége mellett a nyers olvasmányok minőségellenőrzése magában foglalja az olvasási igazítás (olvasási egységesség és GC-tartalom), a számszerűsítés (3′ torzítás, biotípusok és alacsony számok), valamint a reprodukálhatóság (korreláció, fő komponens elemzés és kötegelt hatások) elemzését is.
1. táblázat. Az RNS-seq raw minőségellenőrzési eszközei olvashatók.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Függetlenül attól, hogy rendelkezésre áll-e Genom vagy transzkriptóm referencia, a reads egyedileg térképezhet, vagy hozzárendelhető a referencia több pozíciójához, amelyeket több leképezett olvasásnak vagy többszálúnak neveznek. A genomikus multireads általában ismétlődő szekvenciáknak vagy paralóg gének megosztott doménjeinek köszönhető. A transzkriptóm multi-leképezés gyakrabban fordul elő a gén izoformák miatt. Ezért az átirat-azonosítás és a számszerűsítés fontos kihívást jelent az alternatív módon expresszált gének számára., Ha a referencia nem áll rendelkezésre, RNS-seq olvas össze de novo segítségével csomagok, mint a szappan-transz, oázisok, transz-ABySS, vagy Trinity. PE szál-specifikus, hosszú hosszúságú olvasás előnyösek, mivel sokkal informatívabb. A feltörekvő hosszú olvasású technológiák, mint például a PacBio SMRT szekvenálás és a Nanopore szekvenálás, teljes hosszúságú átiratokat generálhatnak a legtöbb gén számára.
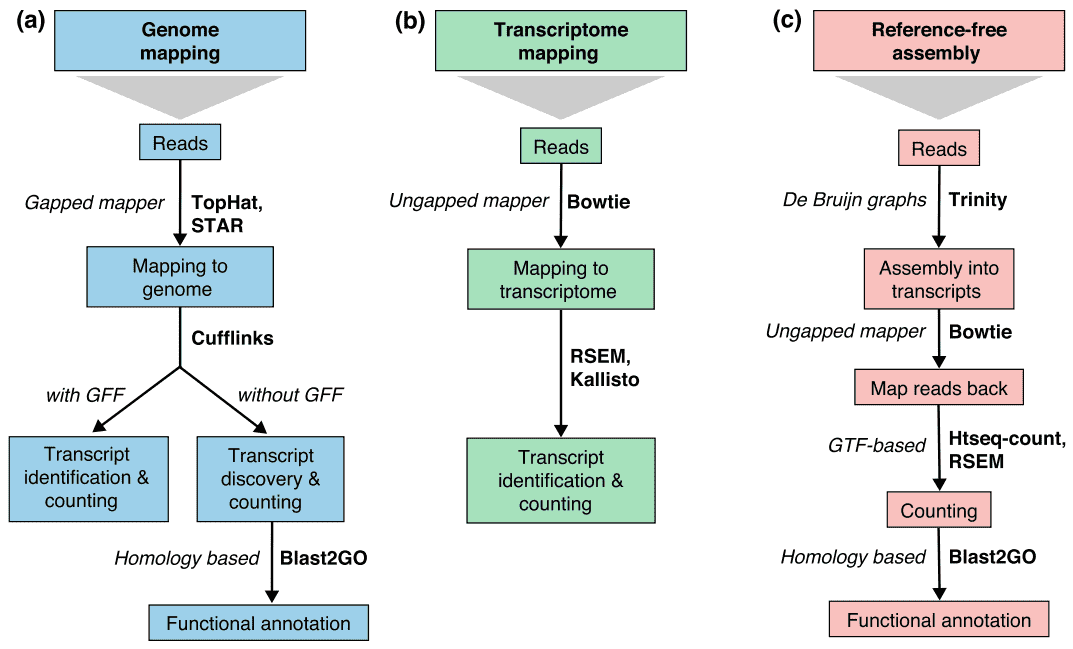
2.ábra. Három alapvető stratégiák RNS-seq olvasási leképezés (Conesa et al. 2016)., Rövidítések: GFF, általános jellemző formátum; GTF, génátviteli formátum; RSEM, RNS-seq elvárások maximalizálásával.
2. táblázat. Az RNS-seq analízis Genom alapú és De novo assembly stratégiáinak összehasonlítása.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Mandzsettagombok | úgy tervezték, hogy kihasználják a PE olvasás, és lehet használni GTF információt azonosítani kifejezett átiratok, vagy infer átiratok de novo a leképezési adatok egyedül. |
| RSEM | A transzkriptóm leképezés kifejezéseinek számszerűsítése. A többszörös leképezés hozzárendelése az átirat és a kimenet között-a mintán belüli normalizált értékek a szekvenálási torzításokhoz korrigálva., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
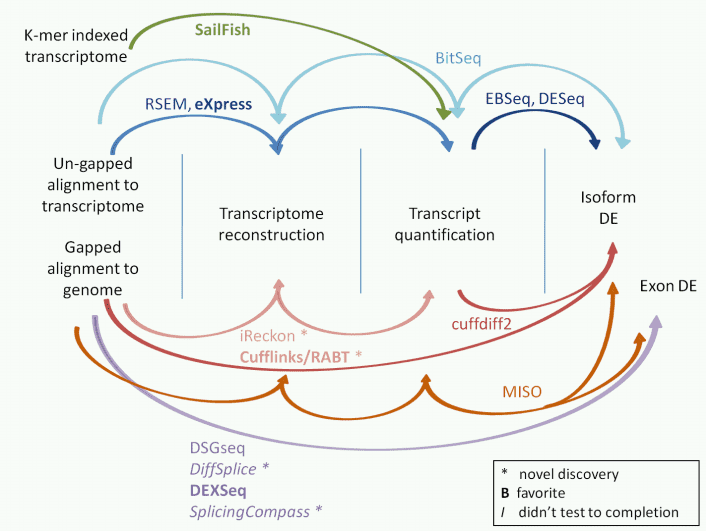
Figure 3. The tools for isoform expression quantification.,
differenciál expressziós vizsgálat
differenciál expressziós vizsgálat annak értékelésére szolgál, hogy az egyik gén különbözik-e az egyik állapotban a másikhoz képest. A különböző minták összehasonlítása előtt normalizáló módszereket kell alkalmazni. Az RPKM és a TPM normalizálja a legfontosabb tényezőt, a szekvenálási mélységet. A TMM, DESeq és UpperQuartile figyelmen kívül hagyhatja a nagyon változó és / vagy erősen kifejezett jellemzőket., Egyéb tényezők, amelyek zavarják belüli minta összehasonlítása magában transcript hossza, helyzeti torzítások a lefedettség átlagos töredék mérete, valamint a GC-tartalom, amely lehet normalizált által eszközök, mint például a DESeq, edgeR, baySeq, valamint NOISeq. A kötegelt hatások továbbra is jelen lehetnek a normalizálás után, amelyet megfelelő kísérleti tervezéssel minimalizálhatunk, vagy eltávolíthatunk olyan módszerekkel, mint a COMBAT vagy az ARSyN.
5. táblázat. A normalizációs eszközök differenciálexpressziós teszteléshez.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Több bioinformatikai eszközt fejlesztettek ki a kísérleti adatok észlelésére. Ezeknek a detektáló eszközöknek az RNS-seq adatokkal történő összehasonlítását a Ding végezte 2017-ben, az eredményeket a 7.táblázat mutatja. Bebizonyították, hogy a TopHat és downstream eszköze, a FineSplice a leggyorsabb eszköz, míg a PASTA a leglassabb program. Ezenkívül az AltEventFinder képes észlelni a legtöbb csomópontot,az RSR pedig a legkisebb számú csomópontot. Más eszközök, például a TopHat, valószínűleg hamis pozitívokat észlelnek., A két eszköz közül, amelyek különbözőképpen illesztett izoformákat észlelnek, az rMATS gyorsabb, mint az rSeqDiff, de kevésbé különbözőképpen illesztett izoformákat észlel, mint az rSeqDiff.
7. táblázat. Kimutatott típusok vagy differenciálisan illesztett izoformák ezen eszközök (Ding et al. 2017).,
Megjelenítés
sok a bioinformatikát eszközök a vizualizáció, az RNS-seq adatok, beleértve a genom böngészők, mint például a ReadXplorer, UCSC böngésző, Integratív Genomika Viewer (IGV), Genom Térképek, Tudós, eszközök kifejezetten RNS-seq adatok, mint például a RNAseqViewer, valamint néhány csomagok differenciál génexpresszió elemzés, amely lehetővé teszi a képi megjelenítés, mint a DESeq2, valamint DEXseq a Bioconductor. A csomagok, mint például a CummeRbund és a Sashimi parcellák, vizualizációs-kizárólagos célokra is kifejlesztésre kerültek.,
funkcionális profilozás
a standard transzkriptomikai vizsgálat legújabb lépése általában azon molekuláris funkciók vagy utak jellemzése, amelyekben a differenciálisan expresszált gének részt vesznek. A gén ontológia, a Bioconductor, a DAVID vagy a Babelomics a legtöbb modellfajra vonatkozó jegyzetadatokat tartalmaz, amelyek funkcionális annotációhoz használhatók. Ami az új átiratokat illeti, a fehérjekódoló átiratok funkcionálisan kommentálhatók ortológiával olyan adatbázisok segítségével, mint a SwissProt, a Pfam és az InterPro., A Gene Ontology (GO) lehetővé teszi a funkcionális információk kicserélhetőségét az ortológusok között. Blast2GO egy népszerű eszköz, amely lehetővé teszi, hogy hatalmas annotáció teljes transzkriptóma ellen a különböző adatbázisok és ellenőrzött szótárak. Az Rfam adatbázis a leginkább jól jellemzett RNS családokat tartalmazza, amelyek hosszú, nem kódoló RNS-ek funkcionális annotációjára használhatók.
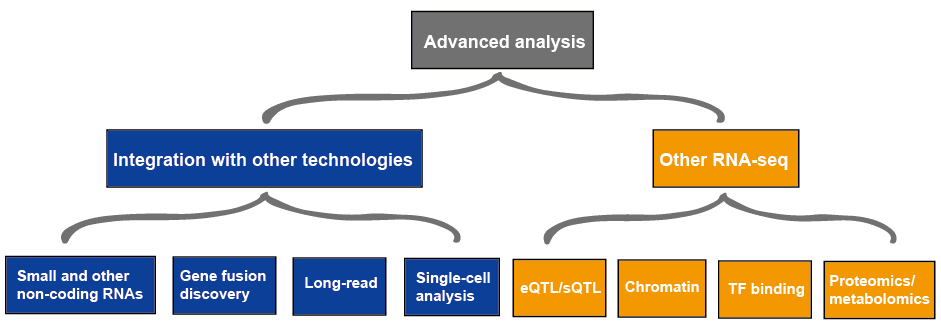
Advanced analysis
az RNS-seq fejlett analízise általában más RNS-seq-t és más technológiákkal való integrációt is magában foglal, amelyet a 4.ábra vázol., További információ az RNS-seq alkalmazásáról, kérjük, tekintse meg ezt a cikket az RNS-Seq alkalmazásai.
3. ábra. Az RNS-seq adatok fejlett elemzése.
tapasztalt bioinformatikai tudósaink a fejlett bioinformatikai eszközök felhasználásával foglalkoznak a következő és harmadik generációs szekvenálás által generált számos szekvenciával. Szekvenálási és bioinformatikai szolgáltatásokat nyújtunk genomika, transzkriptomika, epigenomika, mikrobiális genomika, egysejtű szekvenálás és PacBio SMRT szekvenálás céljából.