le séquençage de L’ARN (RNA-seq) a un large éventail d’applications, et il n’y a pas de pipeline optimal pour tous les cas. Nous passons en revue toutes les principales étapes de l’analyse des données ARN-seq, y compris le contrôle de la qualité, l’alignement de lecture, la quantification des niveaux de gènes et de transcriptions, l’expression différentielle des gènes, le profilage fonctionnel et l’analyse avancée. Ils seront discutés plus tard.
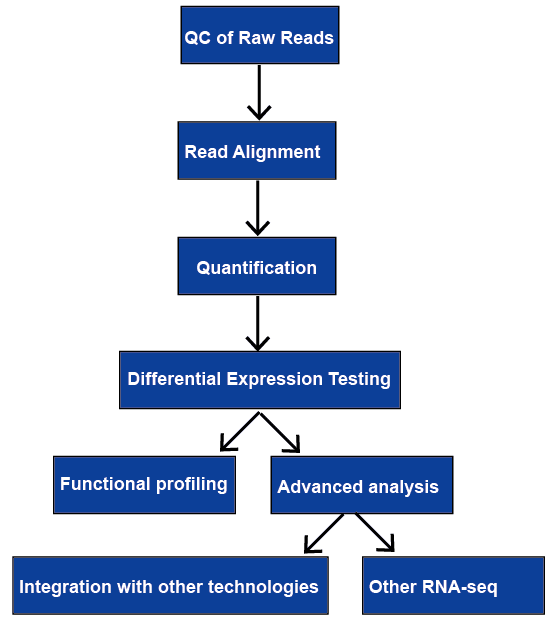
la Figure 1. Le flux de travail général de l’analyse RNA-seq.,
contrôle de la qualité des lectures brutes
Le contrôle de la qualité des lectures brutes ARN-seq consiste en l’analyse de la qualité de la séquence, du contenu en GC, du contenu en Adaptateur, des K-mers surreprésentés et des lectures dupliquées, dédié à la détection des erreurs de séquençage, des contaminations et des artefacts PCR. La qualité de lecture diminue vers la fin des lectures 3’, les bases de faible qualité doivent donc être supprimées pour améliorer la mappabilité., En plus de la qualité des données brutes, le contrôle de la qualité des lectures brutes comprend également l’analyse de l’alignement de lecture (uniformité de lecture et teneur en GC), la quantification (biais 3’, biotypes et faibles dénombrements) et la reproductibilité (corrélation, analyse en composantes principales et effets de lot).
le Tableau 1. Les outils pour le contrôle de la qualité des lectures brutes RNA-seq.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Qu’une référence de génome ou de transcriptome soit disponible, les lectures peuvent être mappées de manière unique ou assignées à plusieurs positions dans la référence, appelées lectures multi-mappées ou multireads. Les multireads génomiques sont généralement dus à des séquences répétitives ou à des domaines partagés de gènes paralogues. Le multi-mappage du Transcriptome se produit plus souvent en raison d’isoformes de gènes. Par conséquent, l’identification et la quantification des transcriptions sont des défis importants pour les gènes exprimés alternativement., Lorsqu’une référence n’est pas disponible, les lectures RNA-seq sont assemblées de novo à l’aide de paquets tels que SOAPdenovo-Trans, Oasis, Trans-ABySS ou Trinity. Les lectures spécifiques au brin PE et de longue Longueur sont préférées car elles sont plus informatives. Les technologies émergentes à lecture longue, telles que le séquençage PacBio SMRT et le séquençage Nanopore, peuvent générer des transcriptions complètes pour la plupart des gènes.
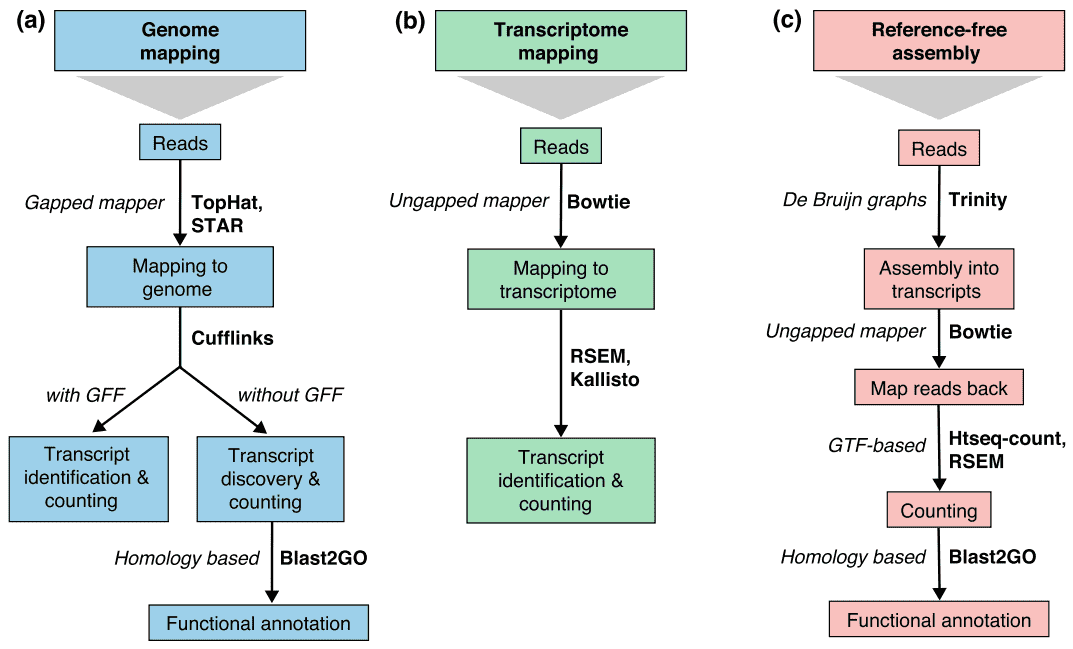
la Figure 2. Trois stratégies de base pour la cartographie de lecture ARN-seq (Conesa et al. 2016)., Abréviations: GFF, format de caractéristique générale; GTF, format de transfert de gène; RSEM, ARN-seq par maximisation des attentes.
le Tableau 2. La comparaison du génome à base et de novo de l’assemblée des stratégies pour l’ARN-seq analyse.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| boutons de manchette | conçus pour tirer parti des lectures PE, et peuvent utiliser les informations GTF pour identifier les transcriptions exprimées, ou peuvent déduire des transcriptions de novo à partir des données de cartographie seules. |
| RSEM | Quantifier l’expression, du transcriptome de la cartographie. allouer des lectures multi-mapping entre les valeurs normalisées de transcription et de sortie dans l’échantillon corrigées pour les biais de séquençage., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
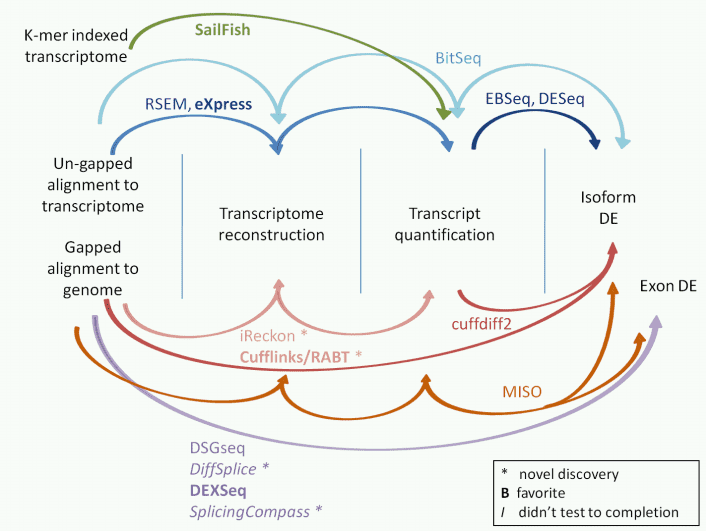
Figure 3. The tools for isoform expression quantification.,
Test D’expression différentielle
le test D’expression différentielle est utilisé pour évaluer si un gène est exprimé de manière différentielle dans une condition par rapport à l’autre. Les méthodes de normalisation doivent être adoptées avant de comparer différents échantillons. RPKM et TPM normalisent le facteur le plus important, la profondeur de séquençage. TMM, DESEQ et UpperQuartile peuvent ignorer des caractéristiques hautement variables et/ou hautement exprimées., D’autres facteurs qui interfèrent avec les comparaisons intra-échantillon impliquent la longueur de la transcription, les biais positionnels dans la couverture, la taille moyenne des fragments et le contenu du GC, qui peuvent être normalisés par des outils tels que DESeq, edgeR, baySeq et NOISeq. Les effets de lot peuvent toujours être présents après la normalisation, ce qui peut être minimisé par une conception expérimentale appropriée, ou supprimé par des méthodes telles que COMBAT ou ARSyN.
le Tableau 5. Les outils de normalisation pour les tests d’expression différentielle.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Plusieurs outils bioinformatiques ont été développés pour détecter à partir de données expérimentales. La comparaison de ces outils de détection à l’aide de données ARN-seq a été réalisée par Ding en 2017, et les résultats sont présentés dans le tableau 7. Ils ont démontré que TopHat et son outil en aval, FineSplice, sont les outils les plus rapides, alors que PASTA est le programme le plus lent. En outre, AltEventFinder peut détecter le plus grand nombre de jonctions, et RSR détecte le plus petit nombre de jonctions. D’autres outils, tels que TopHat, sont susceptibles de détecter les faux positifs., Parmi les deux outils qui détectent les isoformes épissées de manière différentielle, rMATS est plus rapide que rSeqDiff mais détecte moins d’isoformes épissées de manière différentielle que rSeqDiff.
le Tableau 7. Détectées en tant que types ou isoformes épissées de manière différentielle de ces outils (Ding et al. 2017).,
visualisation
Il existe de nombreux outils bioinformatiques pour la visualisation des données ARN-seq, y compris les navigateurs génomiques, tels que ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Genome Maps, Savant, des outils spécialement conçus pour les données ARN-seq, tels que RNAseqViewer, ainsi que certains paquets pour l’analyse différentielle de l’expression des gènes qui Des Packages, tels que des parcelles de CummeRbund et de Sashimi, ont également été développés à des fins exclusives de visualisation.,
profilage fonctionnel
la dernière étape d’une étude transcriptomique standard est généralement la caractérisation des fonctions moléculaires ou des voies dans lesquelles les gènes exprimés différentiellement sont impliqués. Gene Ontology, Bioconductor, DAVID ou Babelomics contiennent des données d’annotation pour la plupart des espèces modèles, qui peuvent être utilisées pour l’annotation fonctionnelle. Comme pour les nouvelles transcriptions, les transcriptions codant des protéines peuvent être annotées fonctionnellement en utilisant l’orthologie à l’aide de bases de données telles que SwissProt, Pfam et InterPro., L’ontologie génique (GO) permet un certain échange d’informations fonctionnelles entre orthologues. Blast2GO est un outil populaire qui permet l’annotation massive de transcriptome complet contre une variété de bases de données et de vocabulaires contrôlés. La base de données Rfam contient la plupart des familles D’ARN bien caractérisées qui peuvent être utilisées pour l’annotation fonctionnelle d’ARN longs non codants.
analyse avancée
l’analyse avancée de L’ARN-seq comprend habituellement d’autres ARN-seq et l’intégration avec d’autres technologies, ce qui est décrit à la Figure 4., Plus d’informations sur les applications de RNA-seq, veuillez consulter cet article Applications de RNA-Seq.
la Figure 3. L’analyse avancée des données ARN-seq.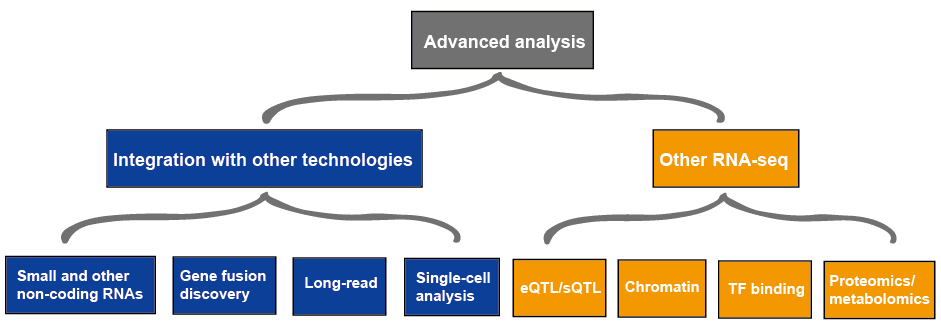
nos scientifiques expérimentés en bioinformatique sont habiles à utiliser les outils de Bioinformatique avancés pour traiter les nombreuses séquences générées par le séquençage de prochaine et troisième génération. Nous fournissons des services de séquençage et de bioinformatique pour la génomique, la transcriptomique, l’épigénomique, la génomique microbienne, le séquençage unicellulaire et le séquençage PacBio SMRT.