Le mois dernier, j’ai été invité à prendre la parole à La Réunion D’analyse et de développement de méthodes du Centers for Mendelian Genomics (CMG) sur « L’estimation basée sur la population de la pénétrance dans les maladies rares”. Voici la version de mon article de blog.
qu’est-ce que la pénétrance et pourquoi faisons-nous des soins?
la pénétrance est la probabilité de développer une maladie particulière compte tenu d’un génotype particulier., On peut parler de pénétrance dépendante de l’âge, donc le pourcentage de personnes ayant le génotype développant la maladie à l’âge de 40 ans, à l’âge de 50 ans, etc.; je parle généralement en termes de risque à vie, ce qui signifie la probabilité que vous développiez la maladie avant de mourir. Inhérent à cela est que, pour les maladies à l’âge adulte, le risque à vie ne peut jamais être tout à fait 100%, parce que vous pourriez toujours mourir d’autre chose en premier.
la pénétrance est extrêmement importante pour les personnes subissant des tests génétiques prédictifs-la première question de nombreuses personnes est: « Est-ce que cela signifie que je vais certainement contracter la maladie?”., Pourtant, il est souvent très difficile d’obtenir une estimation ferme de la pénétrance.
méthodes traditionnelles d’estimation de la pénétrance
Dans un monde idéal, la bonne façon d’estimer la pénétrance serait de vérifier, dès la naissance, une grande cohorte de personnes ayant un génotype particulier, de les suivre jusqu’à ce que toutes soient décédées de quelque chose ou autre, puis de demander combien ont déjà développé la maladie Depuis que la technologie de génotypage a été inventée il y a moins d’une vie humaine, cela n’a jamais été fait pour aucune maladie.
Au Lieu de cela, les chercheurs utilisent souvent des méthodes familiales pour estimer la pénétrance., Une étude typique examinerait tous ceux qui ont été observés avec le génotype donné, et demanderait combien ont diease, ou combien ont la maladie à un certain âge. Les méthodes basées sur la famille souffrent d’un biais de vérification omniprésent ., les familles dans l’étude ont été déterminées sur la base de la présentation de la maladie
à titre d’exemple de ce dernier point, dans la maladie à prions génétiques, seulement 23% des personnes à risque choisissent des tests génétiques prédictifs , et dans les données généalogiques que j’ai eues accès à, nous connaissions les génotypes de seulement 22% des personnes à risque .,
Tous les facteurs énumérés ci-dessus vont dans le même sens, tendant à gonfler l’estimation de la pénétrance.
Les chercheurs sont conscients de ces problèmes depuis longtemps et ont proposé des solutions. Par exemple, la méthode parenté-cohorte consiste à déterminer des individus en bonne santé au hasard d’une population, à les génotyper, à prendre des antécédents familiaux et à comparer les courbes de survie de leurs parents au premier degré., C’est une solution très intelligente, mais elle repose sur la possibilité de déterminer un nombre suffisant de personnes avec un génotype pathogène sans s’assurer de la présence de la maladie. Cela a donc fonctionné pour les variantes BRCA1 et BRCA2 chez les Juifs ashkénazes Américains , mais pour de nombreuses conditions génétiques plus rares, ce n’est pas pratique, car il faudrait recruter des dizaines ou des centaines de milliers de personnes pour trouver ne serait-ce qu’un seul individu avec un génotype d’intérêt.,
méthodes basées sur la population
pour toutes les raisons décrites ci-dessus, il est très utile d’avoir des méthodes orthogonales basées sur la population, pour poser des questions sur la pénétrance. La première idée clé ici est qu’une variante génétique complètement pénétrante ne devrait pas être plus fréquente dans la population que la maladie qu’elle provoque. L’application de cette logique dans la pratique signifie que vous avez besoin de bonnes estimations de la fréquence des allèles, même pour les variantes rares, et cela a été difficile à trouver jusqu’à récemment. ExAC, une base de données de variation génétique dans 60 706 exomes humains, offre de nouvelles opportunités ., De nombreuses personnes dans L’ExAC ont été vérifiées comme cas ou témoins de diverses maladies communes et complexes, mais aucune n’a été vérifiée pour la maladie mendélienne, de sorte que L’ExAC est une bonne base de données de référence pour étudier la plupart des maladies génétiques.
en fournissant des informations sur la fréquence des allèles dans la population générale, ExAC, comme les bases de données de référence antérieures telles que ESP , a clairement montré que la génétique clinique a un gros problème: de nombreuses variantes signalées comme causant une maladie génétique ne causent pas réellement de maladie génétique, ou du moins pas la plupart du temps.,
Deux bases de données — HGMD et ClinVar — recueillent des affirmations de la littérature et des laboratoires cliniques indiquant qu’une variante génétique particulière provoque une maladie génétique particulière. Au dernier décompte, il y avait plus de 100 000 variants génétiques causant des maladies rapportées uniques dans ces bases de données. La personne moyenne à ExAC en a 54 . Évidemment, la personne moyenne n’a pas réellement 54 maladies génétiques., Bien sûr, une grande partie de cet excès est causée par un petit nombre de variantes à très haute fréquence qui ne causent évidemment aucune maladie génétique, et une grande partie pourrait être des variantes récessives trouvées dans un état hétérozygote dans ExAC. Mais même si nous regardons simplement les variants dans les gènes de la maladie dominante à une fréquence d’allèle de <1%, nous voyons toujours 0,89 variants apparemment pathogènes par personne , et ce n’est clairement pas le cas que ~90% des personnes ont une maladie génétique dominante., Donc, à travers le spectre de fréquence des allèles, il y a beaucoup de variantes apparemment pathogènes qui ne sont pas si pathogènes. Lorsque Anne O’Donnell et moi avons examiné les variantes apparemment pathogènes avec les fréquences d’allèles les plus élevées dans ExAC, et demandé comment ils avaient réussi à être mal classés comme pathogènes, nous avons constaté que la plupart du temps, le problème remontait à un article de la littérature qui avait fait une allégation de pathogénicité basée sur des preuves insuffisantes.
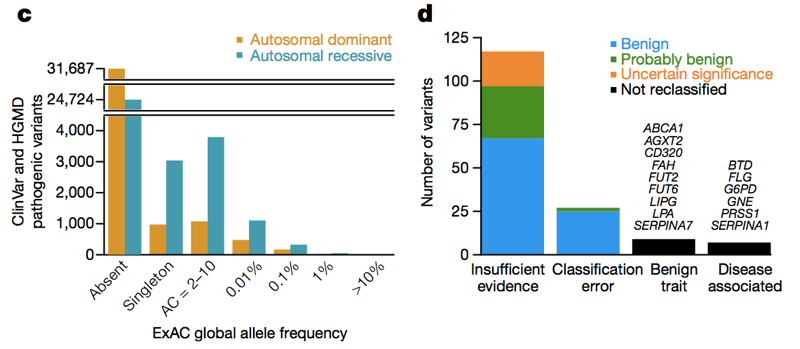
ci-Dessus: les Figures 3C et 3D de ., À travers le spectre de fréquence des allèles et dans les gènes de maladie dominants et récessifs, il y a beaucoup de variantes apparemment pathogènes qui apparaissent dans ExAC. De variants pathogènes à haute fréquence (>1%), quelques-uns sont véritablement pathogènes, certains sont véritablement associés à un trait, mais le trait est bénin, et certains sont des erreurs d’annotation dans les bases de données — mais la majorité sont basées sur la littérature avec des preuves insuffisantes.,
les informations sur la fréquence des allèles de L’ExAC ont maintenant permis de reclasser plus de 200 variants génétiques de pathogène à bénigne, probablement bénigne ou d’importance incertaine . Ces types de reclassements déclenchent parfois le refoulement des Auteurs originaux qui ont proposé qu’une variante provoque une maladie génétique, qui peuvent soutenir qu’une variante pourrait encore être pathogène, mais avec une pénétrance incomplète. Mais comment « incomplet” peut pénétrance incomplète être?, Nous devons être quantitatifs, car si le risque à vie est au plus de 1%, est-il toujours raisonnable de dire qu’une variante « provoque” une maladie génétique ou est « pathogène”? Bien que l’information sur la fréquence des allèles ne puisse jamais prouver qu’une variante n’a pas d’association avec la maladie, elle peut mettre des limites sur ce que pourrait être la pénétrance possible, et dans de nombreux cas, même pour des variantes assez rares, il est possible de montrer qu’il n’y a aucun moyen qu’une variante,
pour obtenir quantitatif, nous devons étendre notre observation antérieure — qu’une variante génétique complètement pénétrante ne devrait pas être plus fréquente dans la population que la maladie qu’elle provoque. Il s’agit de mathématiques simples et de génétique des populations, mais elles ne sont trop souvent pas appliquées dans la pratique. Voici deux façons de penser à la fréquence des allèles lorsque nous faisons des inférences sur la pathogénicité et la pénétrance.
fréquence maximale des allèles crédibles
dites que vous étudiez l’exome d’un patient atteint de la maladie mendélienne et que vous essayez d’identifier la variante causale., Mon collègue James Ware a conçu une stratégie pour filtrer cet exome contre les informations de fréquence des allèles dans ExAC, en tirant parti de la logique suivante., La fréquence maximale des allèles qui est plausible pour qu’une variante cause une maladie génétique dominante est égale à la prévalence de la maladie multipliée par l’hétérogénéité allélique (proportion de cas attribuables à une variante) divisée par pénétrance (moins de variantes pénétrantes peuvent être plus fréquentes), divisée par 2 (parce que nous sommes diploïdes):
\
par exemple, la maladie à prions provoque 1 décès sur 5 000, et la variante la plus commune (E200K) est trouvée dans 5% des cas , donc une variante pénétrante à 100% ne peut pas avoir fréquence d’allèle supérieure à 0,0005% (1 sur 200 000) ., La cardiomyopathie affecte 1 personne sur 500, la variante la plus courante se trouve dans <2% des cas, de sorte qu’une variante pénétrante à 50% ne peut pas avoir une fréquence d’allèle supérieure à 0,004% . La formule pour les maladies récessives est un cran plus compliqué, mais James a également travaillé et il est décrit dans .
alors qu’historiquement les gens ont souvent filtré des variantes avec une fréquence d’allèle>0,1% en essayant d’identifier la cause d’une maladie dominante , nous pouvons en fait être beaucoup plus rigoureux., La mise en garde est qu’à faible nombre d’allèles, notre capacité à estimer la fréquence des allèles est limitée par la variance d’échantillonnage. Par exemple, si nous examinons les variantes observées à une fréquence d’allèle de 1% chez les Européens dans ESP, ces variantes ont également une fréquence d’environ 1% chez les Européens ExAC. Mais les variantes avec une fréquence de 0,1% dans ESP ont tendance à être légèrement plus rares dans ExAC, et la plupart des singletons (variantes vues exactement une fois dans ESP) ne réapparaissent pas une deuxième fois dans ExAC.
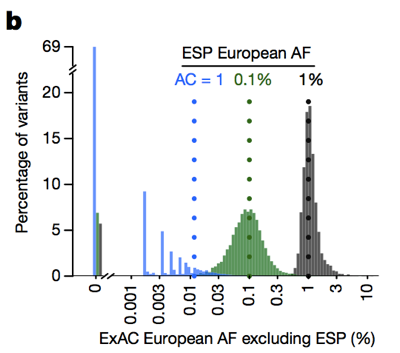
ci-Dessus: Figure 3B de . Plus le nombre d’allèles est faible, moins une estimation de la fréquence des allèles est bonne.,
Par conséquent, plus le nombre d’allèles est faible, plus nous devons être conservateurs. Nous avons conçu un cadre pour le faire en utilisant la limite supérieure de 95% de la distribution de Poisson sur le nombre d’allèles pouvant être observés à une fréquence donnée, et avons des valeurs pré-calculées pour tous les ExAC (disponibles sur FTP) que vous pouvez utiliser — en savoir plus sur les méthodes dans . James a également créé cette application web pratique qui vous permet d’Explorer quelle devrait être la « fréquence maximale des allèles crédibles” pour votre maladie d’intérêt.,
Cette approche est inhérente au fait que plus la pénétrance d’une variante est faible, plus sa fréquence peut être élevée dans la population générale. Mais vous avez également de comprendre que si la pénétrance est assez faible, disons, moins de 10%, alors l’utilité clinique de cette variante est également faible. James et Nicky Whiffin ont présenté des données pour montrer que presque toute l’utilité clinique du séquençage dans la cardiomyopathie provient de variantes avec une fréquence de <0,001% — des variantes plus courantes contribuent cumulativement peu, voire aucun, risque .,
estimation et limites du risque à vie
rappelez-vous que la pénétrance est la probabilité d’une maladie à partir d’un génotype particulier. Ou, si nous considérons un modèle allélique plutôt que génotypique, la probabilité de maladie étant donnée un allèle particulier. Nous pouvons écrire cela comme P (D|A). Une fois que nous l’avons fait, il devient clair que, par le théorème de Bayes,
\
chacun de ces Termes a une signification particulière:
notez ici que « contrôle de la population” désigne un groupe non sélectionné pour la présence, ni pour l’absence, de la maladie. Juste une tranche de la population générale.
Donc:
\
Cette logique n’est rien de nouveau., L’utilisation du théorème de Bayes pour estimer le risque de maladie remonte au moins à l’estimation du risque de cancer chez les fumeurs , et son application à la génétique est considérée depuis presque aussi longtemps . Mais pour que cette équation fonctionne pour les maladies rares, vous avez besoin de très bonnes estimations de la fréquence des allèles de contrôle des cas et de la population, et celles-ci ont été difficiles à trouver jusqu’à récemment. Donc, grâce à ExAC, il y a un nombre croissant de situations où cette équation est pertinente.
Voici le code R que j’ai écrit (à l’origine ici) pour estimer la pénétrance en fonction de cette formule.,
Si vous ne voulez pas exécuter le code R vous-même, James Ware l’a implémenté dans l’onglet « pénétrance” de cette application web afin que vous puissiez simplement brancher vos numéros dans votre navigateur.
afin d’estimer les intervalles de confiance à 95% sur la pénétrance, j’ai adopté l’approche de . Vous entrez le nombre d’allèles (AC) et le nombre d’individus (N) pour les cas et les témoins, et la limite supérieure de L’IC à 95% est calculée sur la base de l’IC supérieur à 95% de la distribution binomiale pour la fréquence des allèles de cas et de l’IC inférieur à 95% pour les témoins., Inversement, la limite inférieure de pénétrance est basée sur la limite inférieure de la fréquence de l’allèle de cas et la limite supérieure de la fréquence de l’allèle de contrôle. Vous pourriez à juste titre ironiser que parce que cette formule utilise 95% CIs sur les deux valeurs de fréquence d’allèle, les intervalles de confiance résultants sont plus grands qu’ils ne devraient l’être. Vous pouvez également ironiser à juste titre que la distribution binomiale n’est pas un bon estimateur à faible nombre d’allèles, en raison du biais illustré à la Figure 3B ci — dessus (et je n’appliquerais certainement jamais cette formule aux singletons-variants observés une seule fois dans ExAC)., Mais à la fin de la journée, pour des raisons dont je parlerai plus près de la fin de ce post, cette formule est vraiment mieux utilisée pour obtenir une estimation approximative de l’ordre de grandeur de la pénétrance. Si vous recherchez une estimation ponctuelle extrêmement précise de la pénétrance, toute cette approche ne fonctionnera probablement pas pour vous de toute façon.
Si vous réorganisez l’équation, une autre façon d’y penser est:
\
cela signifie que le risque accru chez les personnes ayant un génotype est proportionnel au rapport entre le cas et la fréquence des allèles de contrôle de la population., Ainsi, une variante qui augmente le risque de 200 fois devrait être 200 fois plus fréquente parmi les cas que dans la population générale. (Notez que ce rapport des fréquences d’allèles est légèrement différent du rapport de cotes bien que les deux mesures convergent pour des variantes très rares.)
application à la maladie à prions
Nous avons parcouru cette logique dans une étude que nous avons publiée plus tôt cette année, quantifiant la pénétrance des variants de la maladie à prions ., Je me soucie de la maladie à prions pour une raison personnelle — ma femme abrite une variante pathogène dans le PRNP-mais il s’avère que la maladie à prions est également un excellent cas de test pour utiliser la logique ci-dessus pour estimer la pénétrance. Aucun des individus dans ExAC v1 n’a été déterminé sur la maladie neurodégénérative, donc ExAC est vraiment un bon ensemble de données de contrôle de la population pour la maladie à prions. Et parce que les maladies à prions sont « à déclaration obligatoire », les centres de surveillance nationaux ont une détermination des cas exceptionnellement bonne, et grâce à leur générosité dans le partage des données, nous avons pu accumuler un ensemble de données de 10 460 cas séquencés.,
nous avons constaté que les> 60 variants déclarés responsables de la maladie à prions ont cumulativement 52 allèles dans ExAC. Cela signifie que près de 1 personne sur 1 000 a l’une de ces variantes, et donc, ces variantes sont cumulativement beaucoup plus fréquentes que toutes les maladies à prions (qui causent ~1 décès sur 5 000), sans parler de toutes les maladies à prions génétiques (seulement ~15% des cas sont génétiques). Cela suffit pour nous dire que toutes ces variantes ne peuvent éventuellement pas être complètement pénétrantes. Afin de déterminer quelles variantes étaient les coupables, nous avons comparé les séries de cas., Les Variants présentant d’excellentes preuves antérieures de pathogénicité (ségrégation mendélienne et modèles murins) étaient fréquents dans les cas et absents de L’ExAC, ce qui correspond à une pénétrance complète ou presque complète. La plus grande partie du nombre d’allèles en excès dans ExAC a été apportée par des variants qui étaient peu fréquents dans les cas et avaient de faibles preuves antérieures de pathogénicité — ces variants sont probablement bénins ou ne contribuent qu’à un faible risque. Au moins trois variantes sont apparues intermédiaires, car elles étaient trop courantes dans les contrôles pour une pénétrance complète, mais toujours enrichies dans les cas par rapport aux contrôles.,
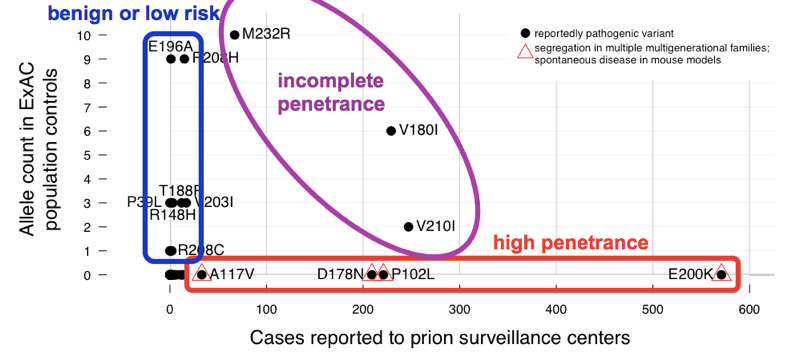
ci-Dessus: une version annotée de la Figure 2 à partir de .
lorsque nous avons estimé la pénétrance pour chaque variante, en utilisant la formule P(D / A) ci-dessus, nous avons constaté qu’il existe tout un spectre de pénétrance pour les variantes PRNP.
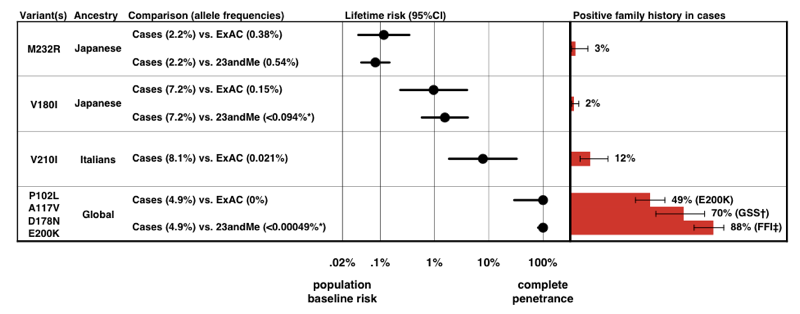
ci-Dessus: Figure 3 de .
notez l’échelle sur l’axe des x — pour une maladie si rare que la probabilité antérieure de la développer n’est que de 0,02%, même une augmentation de 50 fois du risque ne représente que 1% du risque à vie., De manière rassurante, les estimations de pénétrance que nous dérivons de l’information sur la fréquence des allèles seuls concordent assez bien avec la proportion de cas présentant des antécédents familiaux positifs.
Ce travail a déjà conduit à un changement de pronostic pour certaines personnes qui avaient initialement été conseillées qu’elles étaient à risque de variants à forte pénétrance-voir et Erika vérifier L’article de Hayden sur ExAC. Vous pouvez lire mon parcours personnel et celui de Sonia avec cette étude ici.,
application à NR1H3
la sclérose en plaques (SEP) est une maladie complexe avec de nombreux facteurs de risque génétiques , mais aucune forme mendélienne de la maladie n’est connue pour exister. Plus tôt cette année, une étude a rapporté qu’une variante missense dans un récepteur hormonal nucléaire — NR1H3 R415Q — provoque la toute première forme mendélienne de SEP . Cette affirmation était basée sur la ségrégation dominante avec la maladie dans deux familles, mais le score LOD n’était que de 2,2 — en dessous du seuil de signification à l’échelle du génome dans les études de couplage familial, qui ressemble plus à 3,0 ou 3,6 . Et la variante en question a une fréquence d’allèle de 0.,031% en ExAC européens non Finlandais. Cela peut ne pas ressembler à une fréquence d’allèle élevée, mais il s’avère être beaucoup trop élevé pour que cette variante provoque la MS mendélienne .
considérez que la SEP présente un risque à vie (dans la population générale) de 0,25% chez les femmes et de 0,14% chez les hommes . Si 0,06% des personnes de la population générale sont des hétérozygotes R415Q, et si même la moitié d’entre elles développaient la SEP, alors cette variante à elle seule représenterait 0,03% de la population développant la SEP.donc, si un total de 0,25% des personnes développent la SEP, alors environ 12% d’entre elles devraient avoir cette variante., Au lieu de cela, la variante n’a été trouvée que chez 1 individu sur une série de cas de 2 053 patients atteints de SEP .
cela correspond à une fréquence d’allèle de 0,024% dans les cas, ou de 0,049% si nous permettons de compter 2 cas dans la série de cas. Ce n’est pas significativement plus élevé que la fréquence dans ExAC. Mais si cette variante provoque la SEP, elle devrait être plus fréquente dans les cas — beaucoup plus fréquents. Rappelez-vous notre formule réarrangée plus tôt: P(D|A)/P(D) = P(A|D)/P(A). Cela signifie que si une variante augmente le risque de X fois, elle devrait être X fois plus fréquente chez les témoins. Donc, si le risque de base de MS est 0.,25% et cette variante est 50% pénétrant, il devrait être 50/.25 = 200 fois plus fréquent dans les cas que les témoins. S’il avait même une pénétrance de 10%, il devrait toujours être de 10/.25 = 40 fois plus fréquent dans les cas que dans les contrôles. Alternativement, vous pouvez penser en termes de rapports de cotes au lieu de probabilités. Le risque à vie de 0,25% dans la population générale signifie une cote de 1:399, et si R415Q conférait un risque à vie de 50%, ce serait une cote de 50:50. (50/50)/(1/399) = 399, ainsi, le rapport de cotes pour R415Q devrait être de 399 pour que cette variante ait une pénétrance de 50%.,
Au Lieu de cela, si nous appliquons notre formule en utilisant le code R de la précédente, en supposant un risque de référence de 0,25% et en fondant le calcul sur 2 allèles sur 2 053 cas, contre 21 allèles chez 33 369 personnes ExAC, nous constatons que la limite supérieure de L’IC à 95% sur la pénétrance est de 2,2%. Ainsi, même si le R415Q était associé au risque de SEP, il ne pouvait pas conférer plus de 2,2% de risque à vie de développer la SEP .,
dans leur réponse formelle et dans PubMed Commons, les auteurs ont soulevé une comparaison avec LRRK2 G2019S dans la maladie de Parkinson, dont tout le monde convient qu’elle est pathogène, mais qui se trouve également dans ExAC et n’a qu’un rapport de cotes modeste, estimé à 9.6 . Pour cette variante, les mathématiques fonctionne. La maladie de Parkinson est au moins un ordre de grandeur plus répandue que la SEP, avec un risque à vie estimé entre 3,7% et 6,7% . Cet ordre de grandeur plus grande prévalence signifie que l’enrichissement ~10 fois qui a été observé-LRRK2 G2019S se trouve dans environ 0.,1% des contrôles et 1% des cas — est à peu près compatible avec le risque déclaré de Parkinson à vie d’environ 32% conféré par cette variante . Ces détails quantitatifs sont importants et sont différents pour chaque variante et chaque maladie. C’est pourquoi les formules discutées dans cet article sont utiles, même si elles ne fournissent que des estimations très approximatives et sont sujettes à plusieurs mises en garde, comme expliqué ci-dessous.
mises en garde
dans les deux applications décrites ci-dessus, l’information sur la fréquence des allèles a été utilisée pour obtenir une estimation approximative de la pénétrance., Dans la maladie à prions, nous avons pu montrer que les variants précédemment présumés fortement pénétrants conféraient un risque à vie plus élevé de l’ordre de 0,1%, 1% ou 10%. Dans L’histoire NR1H3, les informations sur la fréquence des allèles étaient suffisantes pour montrer que la variante prétendument causale ne pouvait pas conférer plus de quelques pour cent de risque à vie.
mais essayer d’utiliser les données de fréquence des allèles pour obtenir une estimation plus serrée de la pénétrance serait très difficile. Par exemple, les études familiales ont été en désaccord sur la pénétrance du PRNP E200K, avec des estimations allant de 60% à 90% de risque à vie ., Depuis la sortie de l’étude sur les prions, quelques personnes de familles E200K me demandent si les données ExAC peuvent aider à déterminer où le risque se situe dans cette plage. La réponse est malheureusement impossible.
Voici les raisons les plus importantes pour lesquelles je pense que toutes les estimations de pénétrance basées sur la fréquence des allèles doivent être prises avec un grain de sel:
- Si une variante est très pénétrante, il est difficile d’obtenir une série de CAs qui ne contient pas d’individus apparentés. Si votre série de cas est liée, vous n’avez techniquement pas d’estimation impartiale de P (A / D).,
- Si une maladie est mortelle, il est difficile d’obtenir une série de contrôle de la population qui n’est pas au moins quelque peu épuisée parmi les personnes ayant des variantes qui causent cette maladie. Donc, vous n’avez pas non plus d’estimation impartiale de P(A).
- Les comparaisons de la fréquence des allèles cas et témoins sont vulnérables à la confusion par stratification de la population. Dans l’étude sur les prions, nous n’avions pas de données SNP à l’échelle du génome sur les cas, il n’y avait donc aucun moyen de contrôler parfaitement cela.,
- De nombreuses variantes causales des maladies rares sont si rares que même avec ExAC, nous n’avons pas encore d’estimations suffisamment précises de la fréquence des allèles pour donner mieux qu’une réponse approximative.
cela étant dit, l’estimation de la fréquence des allèles basée sur la population est toujours un bon moyen d’obtenir des estimations approximatives de l’ordre de grandeur de la pénétrance et d’effectuer des vérifications de la santé mentale pour savoir si une variante génétique pourrait être causale d’une maladie rare.