El mes pasado fui invitado a hablar en la reunión de análisis y desarrollo de métodos de los Centros para la genómica mendeliana (CMG) sobre «estimación poblacional de penetrancia en enfermedades raras». Aquí está la versión del blog de mi charla.
¿qué es la penetrancia y por qué nos importa?
penetrancia es la probabilidad de desarrollar una enfermedad particular dado un genotipo particular., Se puede hablar de penetrancia dependiente de la edad, por lo que el porcentaje de personas con el genotipo que desarrollan la enfermedad a los 40 años, a los 50 años, y así sucesivamente; por lo general hablo en términos de riesgo de por vida, es decir, la probabilidad de que alguna vez desarrollar la enfermedad antes de morir. Inherente a esto es que, para las enfermedades de aparición adulta, el riesgo de por vida nunca puede ser del todo 100%, porque siempre podría morir de otra cosa primero.
La penetrancia es muy importante para las personas que se someten a pruebas genéticas predictivas: la primera pregunta de muchas personas es: «¿esto significa que definitivamente contraeré la enfermedad?”., Sin embargo, a menudo es muy difícil llegar a una estimación firme de penetrancia.
métodos tradicionales para estimar la penetrancia
en un mundo ideal, la forma correcta de estimar la penetrancia sería determinar, desde el nacimiento, una gran cohorte de personas con un genotipo particular, seguirlas hasta que todas hayan muerto de algo u otro, y luego preguntar cuántos desarrollaron la enfermedad antes de morir. Desde que la tecnología de genotipado se inventó hace menos de una vida humana, esto nunca se ha hecho para ninguna enfermedad.
en su lugar, los investigadores a menudo utilizan métodos basados en la familia para estimar la penetrancia., Un estudio típico observaría a todos los que han sido observados con el genotipo dado, y preguntaría cuántos tienen enfermedad o cuántos tienen enfermedad a cierta edad. Los métodos basados en la familia sufren un sesgo generalizado de determinación ., las familias utilizadas originalmente para establecer que la variante causa la enfermedad se incluyen en el análisis
como ejemplo de este último punto, en la enfermedad priónica genética, solo el 23% de las personas en riesgo eligen pruebas genéticas predictivas , y en los datos de pedigrí que he tenido acceso a, conocíamos los genotipos de solo el 22% de los individuos en riesgo .,
todos los factores enumerados anteriormente trabajan en la misma dirección, tendiendo a inflar la estimación de penetrancia.
Los investigadores han sido conscientes de estos problemas durante mucho tiempo, y han propuesto algunas soluciones. Por ejemplo, el método kin-cohort implica determinar individuos sanos aleatoriamente de una población, genotiparlos, tomar una historia familiar y comparar las curvas de supervivencia de sus parientes de primer grado., Esta es una solución muy inteligente, pero se basa en ser capaz de determinar un número suficientemente grande de personas con un genotipo que causa una enfermedad sin determinar la presencia de la enfermedad. Así que funcionó para las variantes BRCA1 y BRCA2 en judíos asquenazíes estadounidenses, pero para muchas condiciones genéticas más raras, no es práctico, porque se necesitaría reclutar decenas o cientos de miles de personas para encontrar incluso un individuo con un genotipo de interés.,
métodos basados en la población
por todas las razones descritas anteriormente, es muy útil tener métodos ortogonales, basados en la población, para hacer preguntas sobre la penetrancia. La primera idea clave aquí es que una variante genética completamente penetrante no debe ser más común en la población que la enfermedad que causa. Aplicar esta lógica en la práctica significa que necesitas buenas estimaciones de la frecuencia de los alelos incluso para Variantes poco comunes, y eso ha sido difícil de conseguir hasta hace poco. ExAC, una base de datos de variación genética en 60.706 exomas humanos, ofrece nuevas oportunidades ., Muchos individuos en el ExAC se determinaron como casos o controles para varias enfermedades comunes y complejas, pero ninguno se determinó para la enfermedad de Mendel, por lo que el ExAC es una buena base de datos de referencia para estudiar la mayoría de las enfermedades genéticas.
al proporcionar información sobre la frecuencia de los alelos en la población general, ExAC, al igual que las bases de datos de referencia anteriores , como ESP, ha dejado claro que la genética clínica tiene un gran problema: muchas variantes que causan enfermedades genéticas no causan enfermedades genéticas, o al menos no la mayoría de las veces.,
dos bases de datos-HGMD y ClinVar-recogen afirmaciones de la literatura y de los laboratorios clínicos que indican que una variante genética particular causa una enfermedad genética particular. En el último recuento, había más de 100,000 variantes genéticas únicas causantes de enfermedades reportedy en estas bases de datos. La persona promedio en ExAC tiene 54 de ellos . Obviamente, la persona promedio no tiene 54 enfermedades genéticas., Por supuesto, gran parte de este exceso es causado por un pequeño número de variantes de muy alta frecuencia que obviamente no causan ninguna enfermedad genética, y gran parte de ella puede ser, según se informa, variantes recesivas que se encuentran en un estado heterocigoto en ExAC. Pero incluso si nos limitamos a mirar las variantes en los genes de la enfermedad dominante en una frecuencia alélica de < 1%, todavía vemos 0.89 variantes patógenas por persona, y claramente no es el caso que ~90% de las personas tienen una enfermedad genética dominante., Así que a través del espectro de frecuencias alélicas, hay muchas variantes patógenas que no son tan patógenas. Cuando Anne O’Donnell y yo miramos las variantes supuestamente patógenas con las frecuencias alélicas más altas en ExAC, y preguntamos cómo habían logrado clasificarse erróneamente como patógenas, encontramos que la mayoría de las veces el problema se remonta a un artículo en la literatura que había hecho una afirmación de patogenicidad basada en evidencia insuficiente.
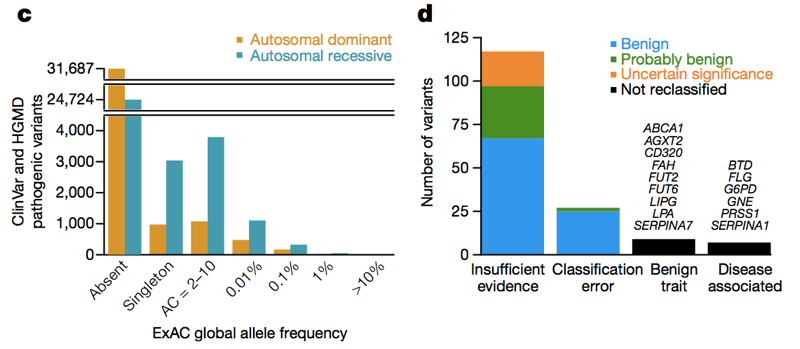
Arriba: las Figuras 3C y 3D ., A través del espectro de frecuencias alélicas y en ambos genes de enfermedad dominante y recesiva, hay una gran cantidad de variantes patógenas que aparecen en ExAC. De alta frecuencia (>1%) variantes patógenas, algunas son genuinamente patógenas, algunas están genuinamente asociadas a rasgos, pero el rasgo es benigno, y algunas son errores de anotación en bases de datos, pero la mayoría se basan en la literatura con evidencia insuficiente.,
la información sobre la frecuencia de los alelos de ExAC ha permitido reclasificar más de 200 variantes genéticas de patógenas a benignas, probablemente benignas o de significación incierta . Este tipo de reclasificaciones a veces desencadenan un retroceso de los autores originales que propusieron que una variante causa una enfermedad genética, que pueden argumentar que una variante todavía podría ser patógena, pero con penetrancia incompleta. Pero, ¿cuán «incompleto» puede ser la penetrancia incompleta?, Necesitamos ser cuantitativos, porque si el riesgo a lo largo de la vida es como máximo del 1%, ¿Sigue siendo razonable decir que una variante «causa» una enfermedad genética, o es «patógena»? Si bien la información sobre la frecuencia de los alelos nunca puede demostrar que una variante no tiene asociación con la enfermedad, puede poner límites a lo que podría ser la posible penetrancia, y en muchos casos, incluso para Variantes bastante raras, es posible demostrar que no hay manera de que una variante confiera un nivel de riesgo remotamente cercano al 100%.,
para ser cuantitativo, necesitamos ampliar nuestra observación anterior: que una variante genética completamente penetrante no debería ser más común en la población que la enfermedad que causa. Todo esto es simple matemática y genética de poblaciones, pero con demasiada frecuencia no se aplica en la práctica. Aquí hay dos maneras en que podemos pensar en la frecuencia de los alelos al hacer inferencias sobre patogenicidad y penetrancia.
frecuencia máxima creíble del alelo
digamos que está estudiando el exoma de un paciente con enfermedad de Mendel e intentando identificar la variante causal., Mi colega James Ware ha ideado una estrategia para filtrar ese exoma contra la información de frecuencia alélica en ExAC, aprovechando la siguiente lógica., La frecuencia máxima alélica que es plausible para que una variante cause una enfermedad genética dominante es igual a la prevalencia de la enfermedad por la heterogeneidad alélica (proporción de casos atribuibles a una variante) dividida por penetrancia (variantes menos penetrantes pueden ser más comunes), dividida por 2 (porque somos diploides):
\
por ejemplo, la enfermedad priónica causa 1 de cada 5.000 muertes, y la variante más común (e200k) se encuentra en el 5% de los casos , por lo que una variante penetrante al 100% no puede tener alelo frecuencia superior al 0,0005% (1 de cada 200.000) ., La miocardiopatía afecta a 1 de cada 500 personas, la variante más común se encuentra en <2% de los casos, por lo que una variante penetrante del 50% no puede tener una frecuencia alélica superior al 0,004% . La fórmula para las enfermedades recesivas es una muesca más complicada, pero James también lo ha resuelto y se describe en .
así que mientras que históricamente las personas a menudo han filtrado variantes con una frecuencia alélica > 0.1% al tratar de identificar la causa de una enfermedad dominante , en realidad podemos ser mucho más estrictos., La advertencia es que en los recuentos bajos de alelos, nuestra capacidad para estimar la frecuencia de los alelos está limitada por la varianza de muestreo. Por ejemplo, si nos fijamos en las variantes observadas con una frecuencia alélica del 1% entre los europeos en ESP, estas variantes también tienen una frecuencia de aproximadamente el 1% entre los europeos ExAC. Pero las variantes con una frecuencia de 0.1% en ESP tienden a ser ligeramente más raras en ExAC, y la mayoría de los singletons (variantes que se ven exactamente una vez en ESP) no reaparecen una segunda vez en ExAC.
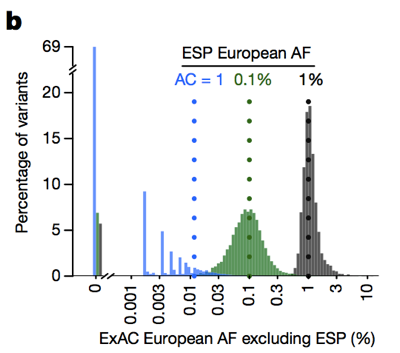
Arriba: Figura 3B desde . Cuanto menor sea el recuento de alelos, menos buena será la estimación de la frecuencia de los alelos que proporciona.,
por lo tanto, cuanto menor sea el recuento de alelos, más conservadores necesitamos ser. Hemos ideado un marco para hacer esto usando el límite superior del 95% de la distribución de Poisson sobre cuántos alelos se pueden observar en una frecuencia dada, y tenemos valores pre-calculados para todo ExAC (disponible en FTP) que puede usar — lea más sobre los métodos en . James también ha creado esta práctica aplicación web que le permite explorar cuál debe ser la «frecuencia alélica máxima creíble» para su enfermedad de interés.,
inherente a este enfoque es que cuanto menor sea la penetrancia de una variante, mayor será la frecuencia que pueda tener en la población general. Pero también hay que calcular que si la penetrancia es bastante baja, digamos, menos del 10%, entonces la utilidad clínica de esa variante también es baja. James y Nicky Whiffin han presentado datos que muestran que casi toda la utilidad clínica de la secuenciación en cardiomiopatía proviene de variantes con una frecuencia de <0,001% — las variantes más comunes contribuyen acumulativamente poco, si es que alguna, riesgo .,
estimación y límites del riesgo de por vida
recuerde que la penetrancia es la probabilidad de enfermedad dado un genotipo particular. O, si consideramos un modelo alélico en lugar de genotípico, la probabilidad de enfermedad dado un alelo en particular. Podemos escribir esto como P (D|A). Una vez que lo hacemos, queda claro que, por el teorema de Bayes,
\
cada uno de estos Términos tiene un significado particular:
observe aquí que «controles poblacionales» significa un grupo no seleccionado por la presencia, ni por la ausencia, de la enfermedad. Sólo una parte de la población general.
So:
\
esta lógica no es nada nuevo., El uso del teorema de Bayes para estimar el riesgo de enfermedad se remonta al menos a la estimación del riesgo de cáncer en fumadores , y su aplicación a la genética se ha considerado durante casi tanto tiempo . Pero para que esta ecuación funcione para las enfermedades raras, se necesitan estimaciones bastante buenas de la frecuencia de los alelos de control de casos y poblaciones, y esas han sido difíciles de conseguir hasta hace poco. Así que gracias a ExAC, hay un número creciente de situaciones donde esta ecuación es relevante.
Aquí está el código R que he escrito (originalmente aquí) para estimar la penetrancia basada en esta fórmula.,
Si no desea ejecutar el código R usted mismo, James Ware lo ha implementado en la pestaña» penetrancia » de esta aplicación web para que pueda conectar sus números en su navegador.
con el fin de estimar intervalos de confianza del 95% en penetrancia, he adoptado el enfoque de . Se ingresa el recuento de alelos (AC) y el número de individuos (N) para los casos y controles, y el límite superior del IC del 95% se calcula con base en el IC del 95% superior de la distribución binomial para la frecuencia del alelo de los casos y el IC del 95% inferior para los controles., Por el contrario, el límite inferior de penetrancia se basa en el límite inferior de la frecuencia del alelo de caso y el límite superior de la frecuencia del alelo de control. Podría objetar correctamente que debido a que esta fórmula utiliza 95% CIs en ambos valores de frecuencia alélica, los intervalos de confianza resultantes son más grandes de lo que deberían ser. También podría objetar con razón que la distribución binomial no es un buen estimador en recuentos bajos de alelos, debido al sesgo ilustrado en la figura 3B mostrada anteriormente (y ciertamente nunca aplicaría esta fórmula a singletones — variantes observadas solo una vez en ExAC)., Pero al final del día, por razones que voy a discutir más cerca del final de este post, esta fórmula es realmente mejor utilizado para la obtención de un estadio, estimación de orden de magnitud de penetrancia. Si está buscando una estimación puntual extremadamente precisa de penetrancia, este enfoque completo probablemente no funcionará para usted de todos modos.
si reorganiza la ecuación, otra forma de pensarlo es:
\
esto significa que el aumento del riesgo entre las personas con un genotipo es proporcional a la relación de frecuencia del alelo caso a control poblacional., Por lo tanto, una variante que aumenta el riesgo en 200 veces debería ser 200 veces más común entre los casos que en la población general. (Tenga en cuenta que esta relación de frecuencias alélicas es ligeramente diferente de odds ratio, aunque las dos medidas convergen para variantes muy raras.)
aplicación a la enfermedad priónica
recorrimos esta lógica en un estudio que publicamos a principios de este año, cuantificando la penetrancia de las variantes de la enfermedad priónica ., Me preocupo por la enfermedad de priones por una razón personal-mi esposa alberga una variante patógena en PRNP – pero resulta que la enfermedad de priones también es un gran caso de prueba para usar la lógica anterior para estimar la penetrancia. Ninguno de los individuos en ExAC v1 fue determinado en enfermedad neurodegenerativa, por lo que ExAC realmente es un buen conjunto de datos de control de la población para la enfermedad prion. Y debido a que las enfermedades priónicas son «notificables», los Centros Nacionales de vigilancia tienen una determinación de casos excepcionalmente buena, y gracias a su generosidad al compartir datos, pudimos acumular un conjunto de datos de 10,460 casos secuenciados.,
se encontró que las > 60 variantes reportadas para causar enfermedad priónica acumulativamente tienen 52 alelos en ExAC. Eso significa que casi 1 de cada 1,000 personas tiene una de estas variantes, y por lo tanto, estas variantes son acumulativamente mucho más comunes que todas las enfermedades priónicas (que causan ~1 de cada 5,000 muertes), y mucho menos todas las enfermedades priónicas genéticas (solo ~15% de los casos son genéticas). Esto es suficiente para decirnos que no todas estas variantes pueden ser completamente penetrantes. Con el fin de determinar qué variantes fueron los culpables, se comparó con la serie de casos., Las variantes con excelente evidencia previa de patogenicidad (segregación mendeliana y modelos de ratón) fueron comunes en los casos y ausentes del ExAC, consistentes con penetrancia completa o casi completa. La mayor parte del recuento de alelos en exceso en ExAC fue contribuido por variantes que eran poco frecuentes en los casos y tenían evidencia previa débil de patogenicidad — estas variantes son probablemente benignas o contribuyen solo con un riesgo bajo. Al menos tres variantes parecían intermedias, ya que eran demasiado comunes en los controles para la penetrancia completa, pero todavía enriquecidas en los casos sobre los controles.,
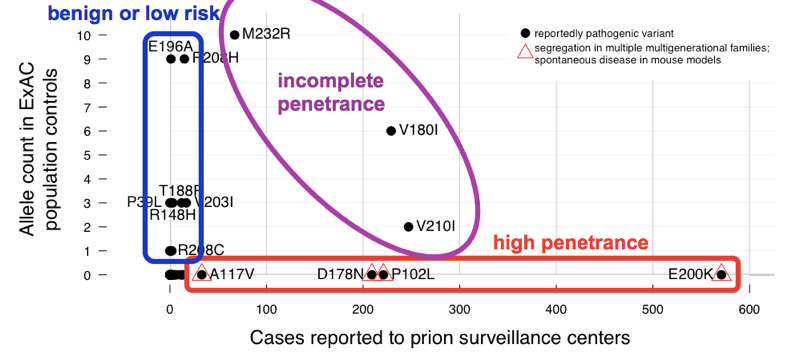
Arriba: una versión anotada de la Figura 2 .
Cuando estimamos la penetrancia para cada variante, usando la fórmula P(D / A) anterior, encontramos que hay un espectro completo de penetrancia para las variantes de PRNP.
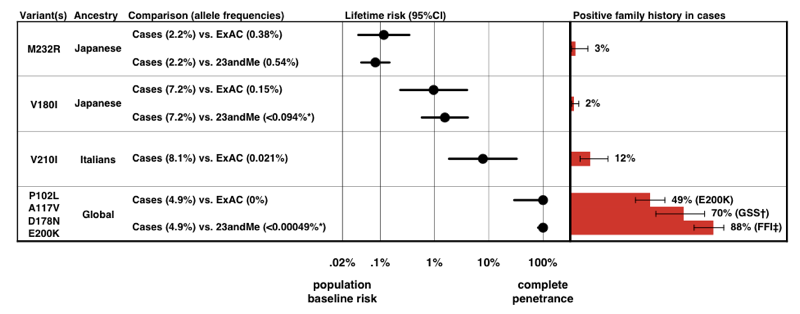
Arriba: Figura 3 desde .
tenga en cuenta la escala en el eje x — para una enfermedad tan rara que la probabilidad previa de desarrollarla es de solo 0,02%, incluso un aumento de 50 veces en el riesgo es de solo 1% riesgo de por vida., De manera tranquilizadora, las estimaciones de penetrancia que derivamos de la información de frecuencia alélica por sí sola concuerdan bastante bien con la proporción de casos que se presentan con una historia familiar positiva.
este trabajo ya ha llevado a un cambio en el pronóstico para algunos individuos que originalmente habían sido aconsejados que estaban en riesgo de variantes de alta penetrancia-ver y Erika comprobar el artículo de Hayden sobre ExAC. Puedes leer mi viaje personal y el de Sonia con este estudio aquí.,
la aplicación a NR1H3
la esclerosis múltiple (EM) es una enfermedad compleja con muchos factores de riesgo genéticos , pero no se sabe que exista una forma mendeliana de la enfermedad. A principios de este año, un estudio informó que una variante missense en un receptor de hormona nuclear — NR1H3 R415Q — causa la primera forma mendeliana de la EM . Esta afirmación se basó en la segregación dominante con enfermedad en dos familias, pero la puntuación de LOD fue solo 2.2 — por debajo del umbral para la importancia de todo el genoma en los estudios de vinculación familiar, que es más como 3.0 o 3.6 . Y la variante en cuestión tiene una frecuencia alélica de 0.,031% en ExAC europeos no finlandeses. Puede que no suene como una frecuencia alélica alta, pero resulta ser demasiado alta para que esta variante cause em mendeliana .
considere que la EM tiene un riesgo de por vida (en la población general) del 0,25% en las mujeres y del 0,14% en los hombres . Si el 0,06% de las personas en la población general son heterocigotos R415Q, y si incluso la mitad de ellos desarrollaron em, entonces esta variante por sí sola representaría el 0,03% de la población que desarrolla em.así que si un total de 0,25% de las personas desarrollan em, entonces alrededor del 12% de ellos deberían tener esta variante., En cambio, la variante solo se encontró en 1 individuo de una serie de casos de 2.053 pacientes con EM .
esto funciona a una frecuencia alélica de 0.024% en casos, o 0.049% si permitimos que 2 casos sean contados en la serie de casos. Esta frecuencia no es significativamente mayor que la de ExAC. Pero si esta variante causa em, debería ser más común en los casos, mucho más común. Recuerde que nuestro reorganizar la fórmula anterior: P(D|A)/P(D) = P(A|D)/P(a). Esto significa que si una variante aumenta el riesgo X veces, debe ser X veces más común en los controles. Así que si el riesgo basal de EM es 0.,25% y esta variante es 50% penetrante, debe ser 50/.25 = 200 veces más común en casos que en controles. Si tuviera un 10% de penetrancia, debería ser 10/.25 = 40 veces más común en los casos que en los controles. Alternativamente, puedes pensar en términos de odds ratios en lugar de probabilidades. El riesgo de 0,25% a lo largo de la vida en la población general significa una probabilidad de 1:399, y si R415Q confiriera un riesgo de 50% a lo largo de la vida, sería una probabilidad de 50:50. (50/50)/(1/399) = 399, por lo tanto, la razón de probabilidades para R415Q tendría que ser 399 para que esta variante tenga una penetrancia del 50%.,
en cambio, si aplicamos nuestra fórmula utilizando el código R de antes, asumiendo un riesgo basal del 0,25% y basando el cálculo en 2 alelos en 2.053 casos, versus 21 alelos en 33.369 individuos ExAC, encontramos que el límite superior del IC del 95% en penetrancia es del 2,2%. Por lo tanto, incluso si R415Q se asociara al riesgo de EM, no podría conferir más de 2.2% de riesgo de por vida de desarrollar EM .,
en su respuesta formal y en PubMed Commons, los autores plantearon una comparación con LRRK2 G2019S en la enfermedad de Parkinson, que todos coinciden en que es patógena, pero que también se encuentra en ExAC y tiene solo una modesta odds ratio, estimada en 9,6 . Para esa variante, las matemáticas funcionan. La enfermedad de Parkinson es al menos un orden de magnitud más prevalente que la EM, con un riesgo de por vida estimado entre el 3,7% y el 6,7% . Este orden de magnitud mayor prevalencia significa que el enriquecimiento ~10 veces que se ha observado-LRRK2 G2019S se encuentra en aproximadamente 0.,1% de los controles y 1% de los casos-es más o menos consistente con el ~32% de riesgo de Parkinson reportado por esta variante . Estos detalles cuantitativos importan, y son diferentes para cada variante y cada enfermedad. Es por eso que las fórmulas discutidas en este post son útiles, a pesar de que solo proporcionan estimaciones muy aproximadas y están sujetas a varias advertencias, como se explica a continuación.
advertencias
en ambas aplicaciones descritas anteriormente, se utilizó información de frecuencia de alelos para obtener una estimación aproximada de la penetrancia., En la enfermedad priónica, pudimos demostrar que las variantes que anteriormente se presumían altamente penetrantes conferían un riesgo de por vida del orden de 0,1%, 1% o 10%. En la historia de NR1H3, la información de frecuencia alélica fue suficiente para mostrar que la variante causal supuestamente no podía conferir más de un porcentaje de riesgo de por vida.
pero tratar de usar datos de frecuencia de alelos para obtener una estimación más ajustada de la penetrancia sería muy difícil. Por ejemplo, los estudios basados en la familia han discrepado sobre la penetrancia de PRNP E200K, con estimaciones que varían de 60% a 90% de riesgo de por vida ., Desde que salió el estudio de prion, algunas personas de familias E200K me preguntaron si los datos de ExAC pueden ayudar a reducir el riesgo dentro de este rango. La respuesta es, desafortunadamente, no puede.
Aquí están las razones más importantes por las que creo que todas las estimaciones de penetrancia basadas en la frecuencia de los alelos deben tomarse con un grano de sal:
- si una variante es altamente penetrante, entonces es difícil obtener una serie de casos que no contenga individuos relacionados. Si su serie de casos tiene relateds, entonces técnicamente no tiene una estimación imparcial de P (A|D).,
- si una enfermedad es fatal, entonces es difícil obtener una serie de control poblacional que no esté al menos un poco agotada de personas con variantes que causan esa enfermedad. Entonces no tienes una estimación imparcial de P(A) tampoco.
- Las comparaciones de frecuencia de alelos de caso y control son vulnerables a la confusión por estratificación poblacional. En el estudio de priones, no teníamos datos de SNP de todo el genoma de los casos, por lo que no había forma de controlar perfectamente esto.,
- muchas variantes causales para enfermedades raras son tan raras que incluso con ExAC, todavía no tenemos estimaciones lo suficientemente precisas de la frecuencia de los alelos para dar mejor que una respuesta aproximada.
con todo lo dicho, la estimación de la frecuencia de los alelos basada en la población sigue siendo una buena manera de obtener estimaciones aproximadas de la penetrancia en orden de magnitud y de realizar controles de cordura sobre si una variante genética podría ser causal plausible para una enfermedad rara.