la secuenciación de RNA (RNA-seq) tiene una amplia gama de aplicaciones, y no hay una canalización óptima para todos los casos. Revisamos todos los pasos principales en el análisis de datos de RNA-seq, incluido el control de calidad, La alineación de lectura, la cuantificación de los niveles de genes y transcripciones, la expresión génica diferencial, el perfil funcional y el análisis avanzado. Se discutirán más adelante.
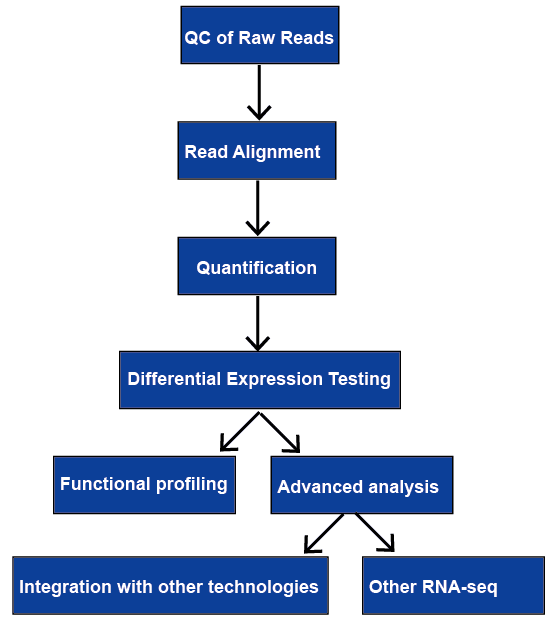
Figura 1. The general workflow of RNA-seq analysis.,
Quality control of raw reads
Quality control of RNA-seq raw reads consists of analysis of sequencing quality, GC content, adaptor content, overrepresented K-mers, and duplicated reads, dedicated to detecting sequencing errors, contaminations, and PCR artifacts. La calidad de lectura disminuye hacia el final de 3′ de lecturas, bases con baja calidad, por lo tanto, deben eliminarse para mejorar la mapabilidad., Además de la calidad de los datos en bruto, el control de calidad de las lecturas en bruto también incluye el análisis de alineación de lectura (uniformidad de lectura y contenido de GC), cuantificación (sesgo de 3′, biotipos y recuentos bajos) y reproducibilidad (correlación, análisis de componentes principales y efectos de lote).
Cuadro 1. Las herramientas para el control de calidad de RNA-seq raw lee.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Independientemente de si una referencia de genoma o transcriptoma está disponible, las lecturas pueden mapearse de forma única o asignarse a múltiples posiciones en la referencia, que se conocen como lecturas multi-mapeadas o multireads. Los multireads genómicos generalmente se deben a secuencias repetitivas o dominios compartidos de genes paralógicos. El multi-mapeo del transcriptoma surge más a menudo debido a las isoformas de los genes. Por lo tanto, la identificación y cuantificación de transcripciones son desafíos importantes para los genes expresados alternativamente., Cuando una referencia no está disponible, las lecturas de RNA-seq se ensamblan de novo utilizando paquetes como SOAPdenovo-Trans, Oases, Trans-ABySS o Trinity. Se prefieren las lecturas específicas de PE y de larga duración, ya que son más informativas. Las tecnologías emergentes de larga lectura, como la secuenciación de PacBio SMRT y la secuenciación de nanoporos, pueden generar transcripciones completas para la mayoría de los genes.
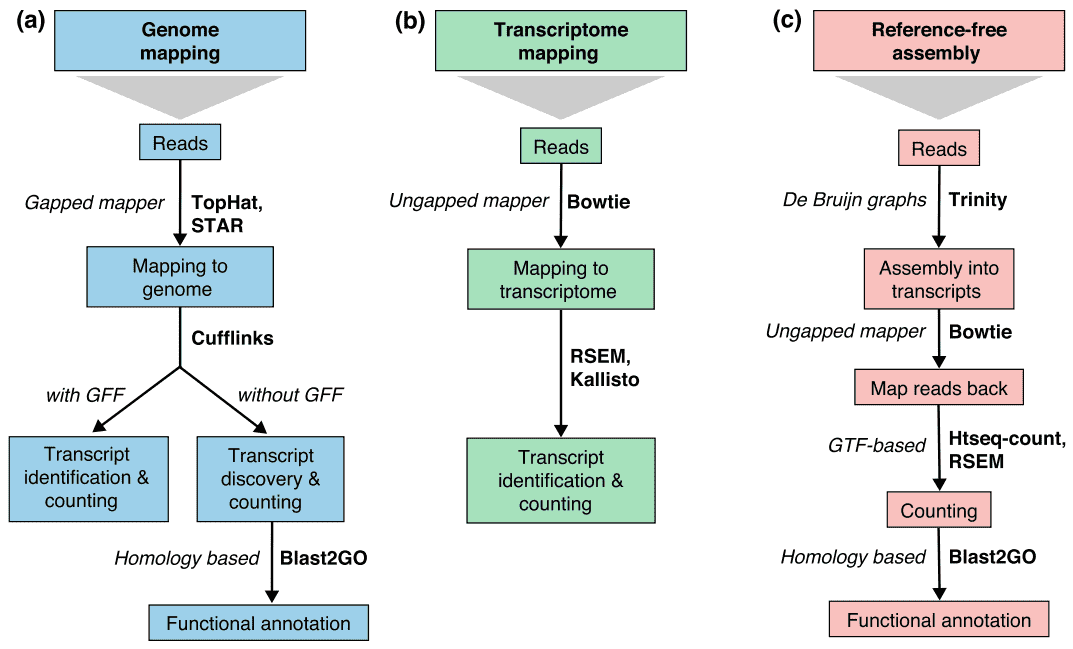
Figura 2. Tres estrategias básicas para el RNA-seq leen mapeo (Conesa et al. 2016)., Abreviaturas: GFF, formato de características generales; GTF, formato de transferencia génica; RSEM, ARN-seq por maximización de expectativa.
Cuadro 2. The comparison of genome-based and de novo assembly strategies for RNA-seq analysis.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Cufflinks | diseñado para aprovechar las lecturas de PE, y puede usar información GTF para identificar transcripciones expresadas, o puede inferir transcripciones de novo solo a partir de los datos de asignación. |
| RSEM | Cuantificar la expresión de la cartografía del transcriptoma.asignar lecturas de asignación múltiple entre transcripción y salida dentro de la muestra valores normalizados corregidos para sesgos de secuenciación., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
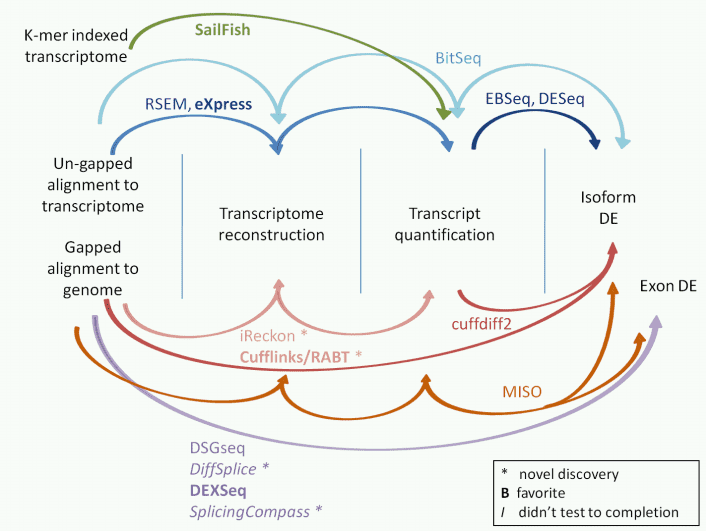
Figure 3. The tools for isoform expression quantification.,
Prueba de expresión diferencial
la prueba de expresión diferencial se utiliza para evaluar si un gen se expresa diferencialmente en una condición en comparación con la otra(s). Es necesario adoptar métodos de normalización antes de comparar diferentes muestras. RPKM y TPM normalizan el factor más importante, la profundidad de secuenciación. TMM, DESeq y UpperQuartile pueden ignorar características altamente variables y/o altamente expresadas., Otros factores que interfieren con las comparaciones intramuestras incluyen la longitud de la transcripción, los sesgos posicionales en la cobertura, el tamaño promedio del fragmento y el contenido de GC, que pueden normalizarse mediante herramientas como DESeq, edgeR, baySeq y NOISeq. Los efectos de lote pueden seguir presentes después de la normalización, que pueden minimizarse mediante un diseño experimental apropiado, o eliminarse mediante métodos como COMBAT o ARSyN.
Cuadro 5. Las herramientas de normalización para pruebas de expresión diferencial.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Se han desarrollado múltiples herramientas bioinformáticas para detectar a partir de datos experimentales. La comparación de estas herramientas de detección utilizando datos de ARN-seq fue realizada por Ding en 2017, y los resultados se muestran en la Tabla 7. Han demostrado que TopHat y su herramienta aguas abajo, Finespplice, son las herramientas más rápidas, mientras que PASTA es el programa más lento. Además, AltEventFinder puede detectar el mayor número de uniones, y RSR detecta el menor número de uniones. Es probable que otras herramientas, como TopHat, detecten falsos positivos., De las dos herramientas que detectan isoformas diferencialmente empalmadas, rMATS es más rápido que rSeqDiff pero detecta menos isoformas diferencialmente empalmadas que rSeqDiff.
cuadro 7. Detectadas como tipos o isoformas diferencialmente empalmadas de estas herramientas(Ding et al. 2017).,
Visualization
Existen muchas herramientas bioinformáticas para la visualización de datos de RNA-seq, incluyendo navegadores genómicos, como ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Genome Maps, Savant, herramientas específicamente diseñadas para datos de RNA-seq, como RNAseqViewer, así como algunos paquetes para el análisis de expresión génica diferencial que permiten la visualización, como DESeq2 y DEXseq en Bioconductor. También se han desarrollado paquetes, como las parcelas de CummeRbund y sashimi, con fines exclusivos de visualización.,
perfil funcional
El último paso en un estudio transcriptómico estándar es generalmente la caracterización de las funciones moleculares o vías en las que los genes expresados diferencialmente están involucrados. La ontología génica, Bioconductor, DAVID O Babelómica contienen datos de anotación para la mayoría de las especies modelo, que se pueden usar para la anotación funcional. En cuanto a las transcripciones nuevas, las transcripciones de codificación de proteínas pueden ser anotadas funcionalmente usando ortología con la ayuda de bases de datos como SwissProt, Pfam e InterPro., La ontología génica (GO) permite cierta intercambiabilidad de información funcional entre ortólogos. Blast2GO es una herramienta popular que permite la anotación masiva de transcriptomas completos contra una variedad de bases de datos y vocabularios controlados. La base de datos Rfam contiene la mayoría de las familias de ARN bien caracterizadas que se pueden utilizar para la anotación funcional de ARN largos no codificantes.
análisis avanzado
el análisis avanzado de ARN-seq generalmente incluye otros ARN-seq e integración con otras tecnologías, lo que se describe en la Figura 4., Más información sobre aplicaciones de RNA-seq, por favor vea este artículo aplicaciones de RNA-Seq.
Figura 3. El análisis avanzado de los datos de RNA-seq.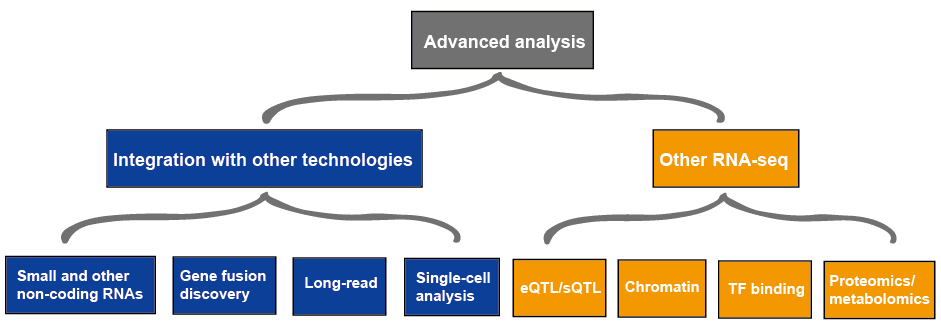
nuestros experimentados científicos bioinformáticos son expertos en la utilización de las herramientas bioinformáticas avanzadas para hacer frente a las numerosas secuencias generadas por la secuenciación de próxima y tercera generación. Proporcionamos servicios de secuenciación y bioinformática para genómica, transcriptómica, epigenómica, genómica microbiana, secuenciación unicelular y secuenciación PacBio SMRT.