i Sidste måned var jeg inviteret til at tale ved Centre for Mendelian Genomics (CMG) Analyse-og metodeudvikling møde om “Befolkning-baseret vurdering af penetrance i sjældne sygdom”. Her er blogindlæg version af my talk.
hvad er penetrans og hvorfor er vi ligeglade?
penetrans er sandsynligheden for at udvikle en bestemt sygdom givet en bestemt genotype., Man kan tale om aldersafhængig penetrans, så procentdelen af mennesker med genotypen, der udvikler sygdommen efter alder 40, efter alder 50, og så videre; jeg taler normalt med hensyn til livstidsrisiko, hvilket betyder sandsynligheden for, at du nogensinde udvikler sygdommen, før du dør. Iboende i dette er, at, for voksne-debut sygdomme, levetid risiko kan aldrig helt være 100%, fordi du altid kunne dø af noget andet først.Penetrance er enormt vigtigt for personer, der gennemgår forudsigelig genetisk test-mange menneskers første spørgsmål er: “betyder det, at jeg helt sikkert får sygdommen?”., Alligevel er det ofte meget vanskeligt at komme med et fast skøn over penetrans.
traditionelle metoder til vurdering af penetrance
I en ideel verden, den rigtige måde at vurdere penetrance ville være at fastslå, fra fødslen, en stor kohorte af personer med en bestemt genotype, følge dem, indtil alle er døde af noget, eller andet, og så spørge, hvor mange, der nogensinde er udviklet sygdommen, før de døde. Da genotypeteknologi blev opfundet for mindre end en menneskelig levetid siden, er dette aldrig blevet gjort for nogen sygdom.
i stedet bruger forskere ofte familiebaserede metoder til at estimere penetrans., En typisk undersøgelse ville se på alle, der er blevet observeret med den givne genotype, og spørge, hvor mange der har sygdom, eller hvor mange der har sygdom i en bestemt alder. Familiebaserede metoder lider af gennemgribende konstateringsbias ., opgøres på grundlag af fremlæggelsen af sygdom
Som et eksempel på dette sidste punkt, i den genetiske prionsygdom, kun 23% på udsatte mennesker vælger prædiktiv gentest , og i stamtavle data, som jeg har haft adgang til, vi kendte genotyper af kun 22% af de udsatte personer .,
alle ovennævnte faktorer arbejder i samme retning og har tendens til at puste ens skøn over penetrering.
forskere har været opmærksomme på disse problemer i lang tid og har foreslået nogle løsninger. Som et eksempel involverer kin-kohortmetoden at konstatere sunde individer tilfældigt fra en befolkning, genotypere dem, tage en familiehistorie og sammenligne overlevelseskurver for deres første graders slægtninge., Dette er en meget smart løsning, men den er afhængig af at være i stand til at konstatere et stort nok antal mennesker med en sygdomsfremkaldende genotype uden at konstatere sygdommens tilstedeværelse. Så det fungerede for BRCA1-og BRCA2-varianter i amerikanske Ashkena .i-jøder , men for mange sjældnere genetiske tilstande er det upraktisk, fordi du bliver nødt til at rekruttere titusinder eller hundreder af tusinder af mennesker for at finde endnu et individ med en genotype af interesse.,
populationsbaserede metoder
af alle de ovenfor beskrevne grunde er det meget nyttigt at have ortogonale, befolkningsbaserede metoder til at stille spørgsmål om penetrans. Den første nøgleindsigt her er, at en fuldstændig gennemtrængende genetisk variant ikke bør være mere almindelig i befolkningen end den sygdom, den forårsager. Anvendelse af denne logik i praksis betyder, at du har brug for gode estimater af allelfrekvens selv for ualmindelige varianter, og det har været svært at komme med indtil for nylig. E .ac, en database med genetisk variation i 60,706 menneskelige e .omer, giver nye muligheder ., Mange individer i E .ac blev konstateret som tilfælde eller kontrol for forskellige almindelige, komplekse sygdomme, men ingen blev konstateret for Mendelian sygdom, så e .ac er en god referencedatabase til undersøgelse af de fleste genetiske sygdomme.
Ved at give allel frekvens oplysninger i den generelle befolkning, ExAC, som tidligere reference databaser som ESP , har gjort det klart, at klinisk genetik har et stort problem: mange varianter rapporteret at forårsage genetiske sygdom faktisk ikke forårsage genetiske sygdomme, eller i det mindste ikke det meste af tiden.,
To databaser — HGMD og ClinVar — saml-påstande fra litteraturen og fra kliniske laboratorier om, at en bestemt genetisk variant, der forårsager en bestemt genetisk sygdom. Ved sidste optælling, der var over 100.000 unikke reportedy sygdomsfremkaldende genetiske varianter i disse databaser. Den gennemsnitlige person i E .ac har 54 af dem . Naturligvis har den gennemsnitlige person faktisk ikke 54 genetiske sygdomme., Selvfølgelig er meget af dette overskud forårsaget af et lille antal vildt højfrekvente varianter, der naturligvis ikke forårsager nogen genetisk sygdom, og meget af det kan angiveligt være recessive varianter, der findes i en Hetero .ygot tilstand i E .ac. Men selv hvis vi blot ser på varianter i dominant sygdom, gener ved en allel frekvens af <1%, ser vi stadig, 0.89 efter sigende patogene varianter per person , og det er helt klart ikke tilfældet, at ~90% af befolkningen har en dominerende genetisk sygdom., Så på tværs af allelfrekvensspektret er der mange angiveligt patogene varianter, der ikke er så patogene. Når Anne O ‘ Donnell, og jeg kiggede til den efter sigende patogene varianter med den højeste allel frekvenser i ExAC, og spurgte, hvordan de havde formået at blive klassificeret som patogene, fandt vi, at de fleste af den tid, problemet spores tilbage til en artikel i den litteratur, der havde gjort et krav sygdomsfremkaldende baseret på utilstrækkelige beviser.
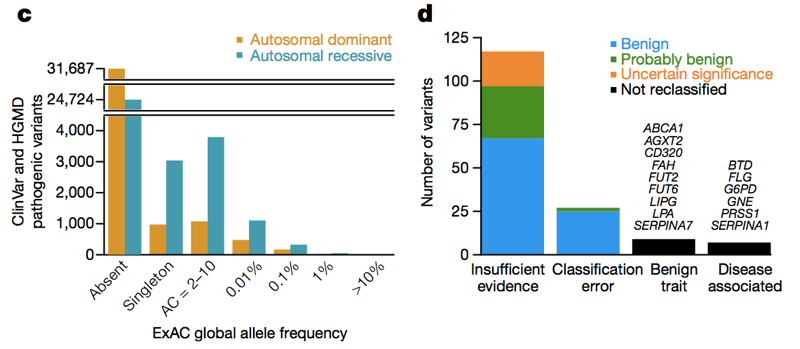
ovenfor: figur 3C og 3D fra ., På tværs af allelfrekvensspektret og i både dominerende og recessive sygdomsgener er der mange angiveligt patogene varianter, der forekommer i E .ac. Af høj (>1%) frekvens efter sigende patogene varianter, et par virkelig er patogene, nogle er virkelig træk-forbundet, men det træk, er godartet, og nogle er fejl i indføring i databaser — men de fleste er baseret på litteratur med utilstrækkelig dokumentation.,
Allelfrekvensinformation Fra e .ac har nu gjort det muligt at omklassificere over 200 genetiske varianter fra patogene til benigne, sandsynligvis godartede eller af usikker betydning . Disse former for omklassifikationer udløser undertiden pushback fra de oprindelige forfattere, der foreslog, at en variant forårsager en genetisk sygdom, som måske hævder, at en variant stadig kunne være patogen, men med ufuldstændig penetrering. Men hvor “ufuldstændig” kan ufuldstændig penetrering være?, Vi er nødt til at blive kvantitative, for hvis livstidsrisiko højst er 1%, er det stadig rimeligt at sige, at en variant “forårsager” en genetisk sygdom eller er “patogen”? Mens allelfrekvensinformation aldrig kan bevise, at en variant ikke har nogen tilknytning til sygdom, kan den sætte grænser for, hvad den mulige penetrering kan være, og i mange tilfælde, selv for temmelig sjældne varianter, er det muligt at vise, at der ikke er nogen måde en variant giver et risikoniveau overalt fjernt tæt på 100%.,
for at blive kvantitativ skal vi udvide vores tidligere observation — at en fuldstændig penetrerende genetisk variant ikke bør være mere almindelig i befolkningen end den sygdom, den forårsager. Dette er alt sammen simpel matematik-og befolkningsgenetik, men det anvendes for ofte ikke i praksis. Her er to måder, vi kan tænke på allelfrekvens, når vi drager konklusioner om patogenicitet og penetrans.
maksimal troværdig allelfrekvens
sig, at du studerer e .omet hos en patient med Mendelian sygdom og forsøger at identificere årsagsvarianten., Min kollega James .are har udtænkt en strategi for filtrering at e .ome mod allel frekvens oplysninger i E .ac, drage fordel af følgende logik., Den maksimale allel frekvens, der er realistisk for en variant til at forårsage en dominerende genetisk sygdom, der er lige til forekomsten af sygdommen gange allel heterogenitet (andelen af sager, der kan henføres til én variant) divideret med penetrance (mindre penetrantprøvning varianter kan være mere almindelige), divideret med 2 (fordi vi er diploide):
\
For eksempel, prionsygdom årsager i 1: 5.000 til dødsfald, og den mest almindelige variant (E200K) er fundet i 5% af tilfældene , så en 100% penetrantprøvning variant kan umuligt have allel frekvens, der er større end 0.0005% (1 ud af 200.000) ., Kardiomyopati påvirker 1 ud af 500 personer, den mest almindelige variant er fundet i <2% af tilfældene, så en 50% penetrantprøvning variant ikke har en allel frekvens, der er større end 0.004% . Formlen for recessive sygdomme er et hak mere kompliceret, men James har også udarbejdet det, og det er beskrevet i .så mens historisk set folk ofte har filtreret ud varianter med en allelfrekvens >0.1% når vi forsøger at identificere årsagen til en dominerende sygdom , kan vi faktisk være meget strengere., Advarslen er, at ved lave alleltællinger er vores evne til at estimere allelfrekvens begrænset af samplingvarians. For eksempel, hvis vi ser på varianter set på et 1% – allel frekvens blandt Europæere i ESP, disse varianter har også omkring en 1% frekvens blandt ExAC Europæere. Men varianter med en 0.1% frekvens i ESP har tendens til at være lidt sjældnere i E .ac, og de fleste enkeltfødte (varianter set nøjagtigt en gang i ESP) vises ikke igen en anden gang i E .ac.
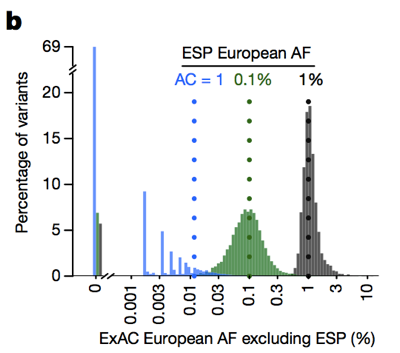
ovenfor: figur 3B fra . Jo lavere allel tæller, jo mindre god et skøn over allel frekvens det giver.,
derfor, jo lavere allel tæller, jo mere konservative skal vi være. Vi har udviklet en ramme for at gøre dette ved hjælp 95% – øvre grænse for Poisson-fordeling på, hvor mange alleler kan iagttages ved en given frekvens, og har pre-beregnede værdier for alle ExAC (findes på FTP), som du kan bruge som du kan læse mere om de metoder, der er i . James har også oprettet denne praktiske appebapp, der giver dig mulighed for at undersøge, hvad den “maksimale troværdige allelfrekvens” skal være for din sygdom af interesse.,
iboende i denne tilgang er, at jo lavere penetrering af en variant er, jo højere frekvens kan den have i den generelle befolkning. Men du skal også regne med, at hvis penetrans er ret lav, siger mindre end 10%, så er den kliniske anvendelighed af denne variant også lav. James og Nicky Whiffin har præsenteret data, der viser, at næsten alle af den kliniske anvendelighed af sekventering i kardiomyopati kommer fra varianter med en frekvens på <0.001% — mere almindelige varianter kumulativt bidrage lidt, hvis nogen risiko .,
estimering og grænser for livstidsrisiko
Husk, at penetrans er sandsynligheden for sygdom givet en bestemt genotype. Eller, hvis vi betragter en allel snarere end genotypisk model, sandsynligheden for sygdom givet en bestemt allel. Vi kan skrive dette som P (D|A). Når vi først har gjort det, bliver det klart, at ved Bayes’ sætning
\
hvert af disse udtryk har en bestemt betydning:
Bemærk her, at “befolkningskontrol” betyder en gruppe, der ikke er valgt for sygdommens tilstedeværelse eller fravær. Bare et udsnit af den generelle befolkning.
så:
\
denne logik er ikke noget nyt., Brugen af Bayes ‘ sætning til at estimere sygdomsrisiko går i det mindste tilbage til estimeringen af risikoen for kræft hos rygere , og dens anvendelse på genetik er blevet overvejet i næsten lige så lang tid . Men for at denne ligning skal fungere for sjældne sygdomme, har du brug for temmelig gode estimater af tilfælde og befolkningskontrol allelfrekvens, og de har været svære at komme med indtil for nylig. Så takket være E .ac er der et stigende antal situationer, hvor denne ligning er relevant.
Her er den R-kode, jeg har skrevet (oprindeligt her) for at estimere penetrans baseret på denne formel.,
Hvis du ikke selv ønsker at køre r-koden, har James .are implementeret den i fanen “penetrance” i denne appebapp, så du bare kan tilslutte dine numre til din bro .ser.
for at estimere 95% konfidensintervaller på penetrans har jeg vedtaget tilgangen til . Du indtaster alleltællingen (AC) og antallet af individer (N) for sager og kontroller, og den øvre grænse for 95% CI beregnes ud fra den øvre 95% CI af Binomialfordelingen for sag allelfrekvens og den nedre 95% CI for kontroller., Omvendt er den nedre grænse for penetrans baseret på den nedre grænse af sag allel frekvens og den øvre grænse af kontrol allel frekvens. Du kan med rette kvæle det, fordi denne formel bruger 95% CIs på begge allelfrekvensværdier, er de resulterende konfidensintervaller større, end de burde være. Du kunne også med rette brokker, at binomialfordelingen er ikke en god estimator ved lave allel, der tæller, på grund af den bias illustreret i Figur 3B, der er vist ovenfor (og jeg har helt sikkert aldrig ville anvende denne formel til enkeltfødte — varianter, der kun observeret en gang i ExAC)., Men i slutningen af dagen, af grunde, jeg vil diskutere tættere på slutningen af dette indlæg, er denne formel virkelig bedst brugt til at opnå en ballpark, størrelsesordens estimat af penetrans. Hvis du leder efter et ekstremt præcist punktestimat af penetrans, fungerer hele denne tilgang sandsynligvis ikke for dig alligevel.
Hvis du omarrangerer ligningen, er en anden måde at tænke på:
\
dette betyder, at den øgede risiko blandt mennesker med en genotype er proportional med forholdet mellem sag og populationskontrol allelfrekvens., Så en variant, der øger risikoen med 200 gange, bør være 200 gange mere almindelig blandt sager end i den generelle befolkning. (Bemærk, at dette forhold mellem allelfrekvenser er lidt anderledes end oddsforholdet, selvom de to mål konvergerer for meget sjældne varianter.)
anvendelse på prionsygdom
Vi gik gennem denne logik i en undersøgelse, vi offentliggjorde tidligere i år, og kvantificerede penetrering af prionsygdomsvarianter ., Jeg er interesseret i prionsygdom af en personlig grund — min kone har en patogen variant i PRNP-men det viser sig, at prionsygdom også er et godt testtilfælde for at bruge ovenstående logik til at estimere penetrans. Ingen af individerne i E .ac v1 blev konstateret på neurodegenerativ sygdom, så e .ac er virkelig et godt datasæt til befolkningskontrol for prionsygdom. Og fordi prionsygdomme er” anmeldelsespligtige”, har nationale overvågningscentre usædvanligt god sagsvurdering, og takket være deres generøsitet i deling af data var vi i stand til at akkumulere et datasæt på 10,460 sekventerede sager.,
Vi fandt, at >60 varianter rapporteret at forårsage prionsygdom kumulativt har 52 alleler i E .ac. Det betyder, at næsten 1 ud af 1.000 mennesker har en af disse varianter, og således, at disse varianter er kumulativt meget mere udbredt end alle prionsygdom (som forårsager ~1 i hver 5000 dødsfald), endsige alle genetiske prionsygdom (kun ~15% af tilfældene er genetisk). Dette er nok til at fortælle os, at ikke alle disse varianter muligvis kan være fuldt gennemtrængende. For at bestemme hvilke varianter der var synderne, sammenlignede vi med case serien., Varianter med fremragende tidligere tegn på patogenicitet (Mendelian segregation og musemodeller) var almindelige i tilfælde og fraværende fra E Exacac, i overensstemmelse med fuldstændig eller næsten fuldstændig penetrering. Det meste af det overskydende allelantal i E .ac blev bidraget med varianter, der var usædvanlige i tilfælde og havde svage tidligere tegn på patogenicitet — disse varianter er sandsynligvis godartede eller bidrager kun med en lav risiko. Mindst tre varianter syntes mellemliggende, da de var for almindelige i kontroller for fuld penetrans, men stadig beriget i tilfælde over kontroller.,
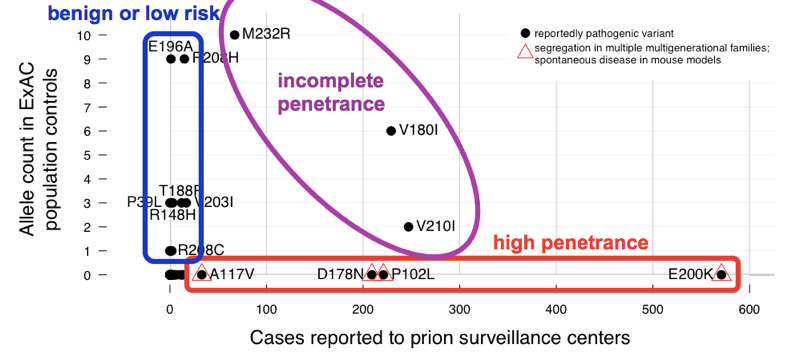
ovenfor: en kommenteret version af figur 2 fra .
Når vi estimerede penetrering for hver variant ved hjælp af P(D / A) – formlen ovenfor, fandt vi, at der er et helt spektrum af penetrering for PRNP-varianter.
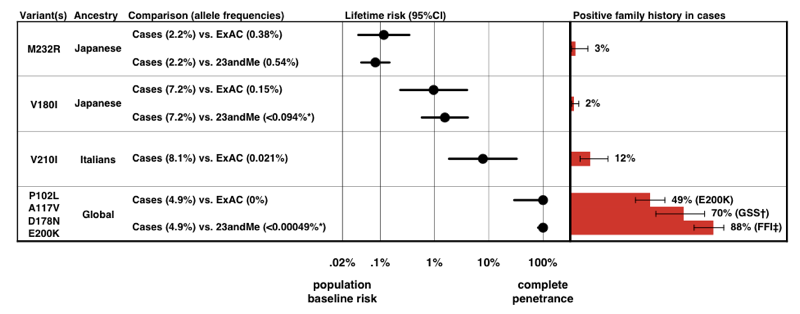
ovenfor: figur 3 fra .
Bemærk skalaen på axis — aksen-for en sygdom, der er så sjælden, at den forudgående Sandsynlighed for at udvikle den kun er 0, 02%, selv en 50 gange stigning i risiko er kun 1% levetidsrisiko., Beroligende estimerer penetransen, at vi stammer fra allelfrekvensinformation alene, stemmer ganske godt med andelen af sager, der er til stede med en positiv familiehistorie.
Dette arbejde har allerede ført til en ændring i prognosen for nogle personer, der havde oprindeligt været rådede til, at de var i risiko for høj-penetrance varianter — se og Erika Check Hayden ‘ s artikel om ExAC. Du kan læse min og Sonias personlige rejse med denne undersøgelse her.,
anvendelse på nr1h3
multipel sklerose (MS) er en kompleks sygdom med mange genetiske risikofaktorer , men det vides ikke, at der findes nogen Mendelsk form af sygdommen. Tidligere i år rapporterede en undersøgelse, at en missense — variant i en nuklear hormonreceptor — NR1H3 R415.-forårsager den første Mendelske form for MS. Denne påstand var baseret på dominerende adskillelse med sygdom i to familier, men den LOD score var kun 2.2 — under tærskelværdien for genome-wide betydning i familie-sammenhæng studier, som er mere som 3.0 eller 3.6 . Og den pågældende variant har en allelfrekvens på 0.,031% i E Exacac ikke-finske europæere. Det lyder måske ikke som en høj allelfrekvens, men det viser sig at være alt for højt for denne variant at forårsage Mendelian MS .
Overvej at MS har en livstidsrisiko (i den generelle befolkning) på 0,25% i kvinder og 0,14% i mænd . Hvis 0,06% af befolkningen i den almindelige befolkning er r415.Hetero .ygoter, og hvis endda halvdelen af dem fortsatte med at udvikle MS, ville denne variant alene tegne sig for 0,03% af befolkningen, der Udvikler MS. så hvis I alt 0,25% af mennesker udvikler MS, så skulle omkring 12% af dem have denne variant., I stedet blev varianten kun fundet i 1 Individuel ud af en case-serie på 2.053 MS-patienter .
Dette fungerer til en allelfrekvens på 0.024% i tilfælde, eller 0.049%, hvis vi tillader, at 2 tilfælde tælles i sagsserien. Dette er ikke signifikant højere end frekvensen i E .ac. Men hvis denne variant forårsager MS, bør det være mere almindeligt i tilfælde-meget mere almindeligt. Husk vores omarrangerede formel tidligere: p(d|a)/p(d) = p(a|d)/p (a). Dette betyder, at hvis en variant øger risikoen med fold-fold, skal den være times gange mere almindelig i kontroller. Så hvis baseline risikoen for MS er 0.,25% og denne variant er 50% penetrant, den skal være 50/.25 = 200 gange mere almindeligt i tilfælde end kontroller. Hvis det endda havde 10% penetrans, skulle det stadig være 10/.25 = 40 gange mere almindelig i tilfælde end i kontroller. Alternativt kan du tænke i forhold til odds nøgletal i stedet for sandsynligheder. Den 0.25% levetid risiko i den almindelige befolkning betyder 1:399 odds, og hvis R415.tillagt 50% levetid risiko, der ville være 50:50 odds. (50/50)/(1/399) = 399, så oddsforholdet for R415.skulle være 399 for at denne variant skal have 50% penetrans.,
i Stedet, hvis vi anvender vores formel ved hjælp af R-kode fra tidligere, forudsat 0.25% baseline risiko og basere beregningen på 2 alleler på 2,053 tilfælde, versus 21 alleler i 33,369 ExAC enkeltpersoner, finder vi, at den øvre grænse af 95%CI på penetrance er 2,2%. Så selvom R415 .var forbundet med MS-risiko, kunne det ikke give mere end 2.2% levetid risiko for at udvikle MS.,
I deres formelle svar, og i PubMed Commons forfatterne rejst en sammenligning til LRRK2 G2019S i Parkinsons sygdom, som alle er enige om er patogene, men der er også fundet i ExAC og har kun et beskedent odds ratio, der er anslået til 9.6 . For den variant fungerer matematikken. Parkinsons sygdom er mindst en størrelsesorden mere udbredt end MS, med livstidsrisiko estimeret overalt fra 3.7% til 6.7% . Denne størrelsesorden større prævalens betyder, at den ~ 10 gange berigelse, der er blevet observeret-LRRK2 g2019s findes i omtrent 0.,1% af kontrollerne og 1% af tilfældene — er omtrent i overensstemmelse med den rapporterede ~32% livstidsrisiko for Parkinson, der er forbundet med denne variant . Disse kvantitative detaljer betyder noget og er forskellige for hver variant og enhver sygdom. Derfor er formlerne, der diskuteres i dette indlæg, nyttige, selvom de kun giver meget grove skøn og er underlagt flere advarsler, som forklaret nedenfor.
advarsler
i begge de ovenfor beskrevne applikationer blev allelfrekvensinformation brugt til at få et groft skøn over penetrans., Ved prionsygdom var vi i stand til at vise, at varianter, der tidligere blev antaget stærkt penetrerende, gav levetids risiko mere i størrelsesordenen 0.1%, 1% eller 10%. I nr1h3-historien var allelfrekvensinformation tilstrækkelig til at vise, at den angiveligt kausale variant ikke kunne give mere end et par procent levetid risiko.men at forsøge at bruge allelfrekvensdata for at få et strammere skøn over penetrans ville være meget udfordrende. For eksempel har familiebaserede undersøgelser uenige om penetrering af PRNP e200k, med estimater fra 60% til 90% livstidsrisiko ., Siden prion-undersøgelsen kom ud, har jeg fået et par mennesker fra e200k-familier til at spørge mig, om e .ac data kan hjælpe med at indsnævre, hvor risikoen er inden for dette interval. Svaret er, desværre, det kan man ikke.
Her er de vigtigste grunde, hvorfor jeg tror, at alle penetrance skøn baseret på allel frekvens skal tages med et gran salt:
- Hvis en variant er meget penetrantprøvning, så er det svært at få en sag serie, der ikke indeholder beslægtede individer. Hvis din sagsserie har relateds, så har du teknisk set ikke et upartisk skøn over P(A|D).,
- hvis en sygdom er dødelig, er det svært at opnå en populationskontrolserie, der ikke i det mindste er noget udtømt af mennesker med varianter, der forårsager denne sygdom. Så så har du heller ikke et upartisk skøn over P(A).
- sammenligninger af tilfælde og kontrol allel frekvens er sårbare over for confounding af befolkning stratificering. I prion-undersøgelsen havde vi ikke genom-brede SNP-data om sager, så der var ingen måde at kontrollere perfekt til dette.,mange årsagsvarianter for sjældne sygdomme er så sjældne, at selv med E .ac har vi endnu ikke tilstrækkeligt præcise estimater af allelfrekvens til at give bedre end et groft svar.
med alt det sagt er befolkningsbaseret allelfrekvensstimering stadig en god måde at få Grove størrelsesordensestimater af penetrans og at udføre sanitetskontrol af, om en genetisk variant sandsynligvis kunne være kausal for en sjælden sygdom.