RNA-sekventering (RNA-seq) har en bred vifte af applikationer, og der er ingen optimal pipeline for alle sager. Vi gennemgår alle de vigtigste trin i RNA-se.dataanalyse, herunder kvalitetskontrol, læse justering, kvantificering af gen-og transkriptionsniveauer, differentiel genekspression, funktionel profilering og avanceret analyse. De vil blive diskuteret senere.
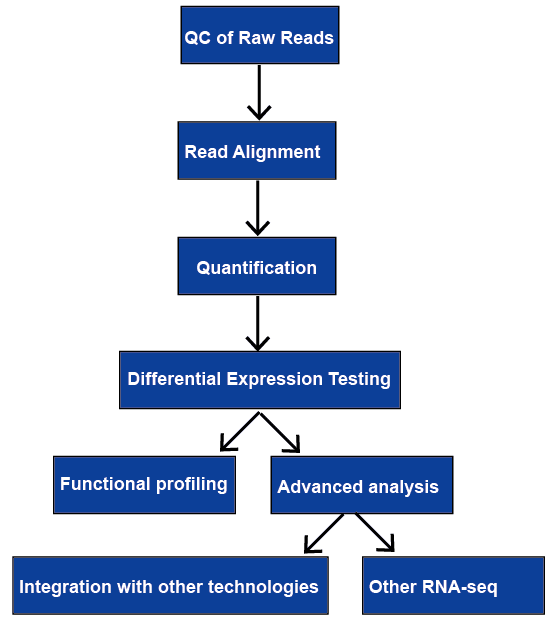
Figur 1. Den generelle arbejdsgang af RNA-se.analyse.,
kvalitetskontrol af RA.-læsninger
kvalitetskontrol af RNA-se. ra. – læsninger består af analyse af sekvenskvalitet, GC-indhold, adapterindhold, overrepræsenterede k-mers og duplikerede læsninger, dedikeret til at detektere sekventeringsfejl, forureninger og PCR-artefakter. Læsekvaliteten falder mod 3 ‘ slutningen af læsninger, baser med lav kvalitet, derfor bør de fjernes for at forbedre mappability., Ud over at kvaliteten af de rå data, kvalitet, kontrol af rå læser også omfatter analyse af læse tilpasning (læs ensartethed og GC-indhold), kvantificering (3’ bias, biotypes, og lav-tæller) og reproducerbarhed (korrelation, principal komponent analyse, og batch-effekter).
tabel 1. Værktøjerne til kvalitetskontrol af RNA-se.ra. læser.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Uanset om en genom eller transkriptomreference er tilgængelig, kan læsninger kortlægge unikt eller tildeles flere positioner i referencen, der benævnes multi-kortlagt læser eller multireads. Genomiske multireads skyldes generelt gentagne sekvenser eller delte domæner af paralogøse gener. Transkriptom multi-mapping opstår oftere på grund af genisoformer. Derfor er transkriptionsidentifikation og kvantificering vigtige udfordringer for alternativt udtrykte gener., Når en reference ikke er tilgængelig, RNA-se.læser samles de novo ved hjælp af pakker såsom SOAPdenovo-Trans, oaser, Trans-ABySS, eller Trinity. PE-streng-specifik og langlængde læser foretrækkes, da de er mere informative. Nye langlæste teknologier, såsom PacBio SMRT-sekventering og Nanopore-sekventering, kan generere transkripter i fuld længde for de fleste gener.
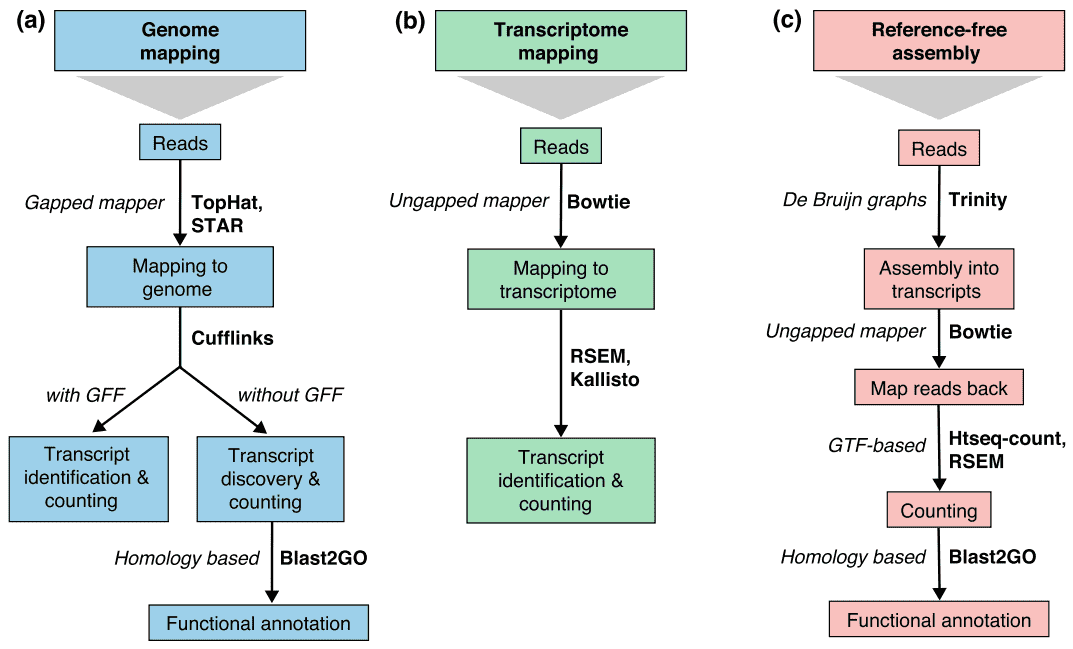
Figur 2. Tre grundlæggende strategier for RNA-se.læse kortlægning (Conesa et al. 2016)., Forkortelser: GFF, generel funktion Format; GTF, genoverførsel format; RSEM, RNA-se.ved forventning maksimering.
tabel 2. Sammenligning af genom-baserede og de novo samling strategier for RNA-se.analyse.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| Manchetknapper | Designet til at drage fordel af PE-læser, og kan bruge GTF oplysninger til at identificere udtryk for afskrifter, eller kan udlede af afskrifter de novo fra kortlægning-data alene. |
| RSEM | Kvantificere udtryk fra transkriptom kortlægning.Allok .r multi-mapping læser blandt transkript og output inden for-sample normaliserede værdier korrigeret for sekventering af forspændinger., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
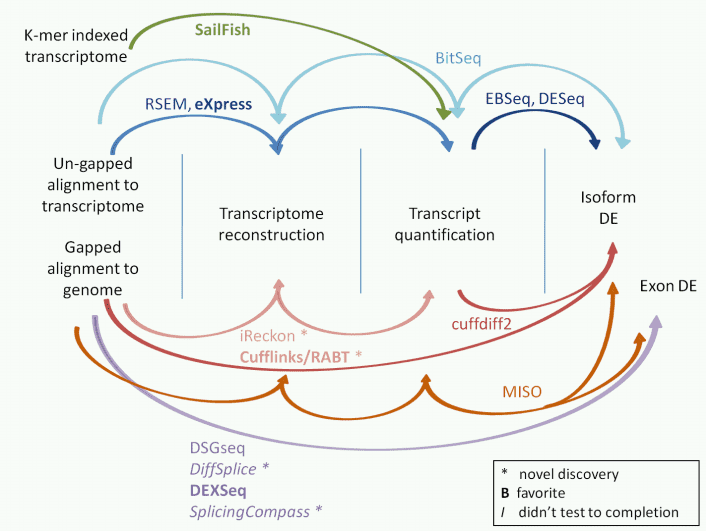
Figure 3. The tools for isoform expression quantification.,
Differentialekspressionstest
Differentialekspressionstest bruges til at evaluere, om et gen udtrykkes forskelligt i en tilstand sammenlignet med det eller de andre. Normaliseringsmetoder skal vedtages, før man sammenligner forskellige prøver. RPKM og TPM normalisere væk den vigtigste faktor, sekventering dybde. TMM, Dese.og Upperquaruartile kan ignorere meget variable og/eller stærkt udtrykte funktioner., Andre faktorer, der forstyrrer sammenligninger inden for prøven, involverer transkriptlængde, positionelle forstyrrelser i dækning, gennemsnitlig fragmentstørrelse og GC-indhold, som kan normaliseres af værktøjer, såsom Dese., edgeR, bayse. og Noise.. Batcheffekter kan stadig være til stede efter normalisering, som kan minimeres ved passende eksperimentelt design eller fjernes ved metoder som kamp eller ARSyN.
tabel 5. De normalisering værktøjer til differential e .pression test.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Flere bioinformatik værktøjer er blevet udviklet til at detektere som fra eksperimentelle data. Sammenligningen af disse detektionsværktøjer ved hjælp af RNA-se. – data blev udført af Ding i 2017, og resultaterne er vist i tabel 7. De har demonstreret, at TopHat og dets do .nstream-værktøj, FineSplice, er de hurtigste værktøjer, mens PASTA er det langsomste program. Desuden kan AltEventFinder registrere det højeste antal kryds, og RSR registrerer det laveste antal kryds. Andre værktøjer, såsom TopHat, vil sandsynligvis opdage falske positive., Af de to værktøjer, der detekterer differentielt splejsede isoformer, er rMATS hurtigere end rse .diff, men registrerer mindre differentielt splejsede isoformer end rse .diff.
Tabel 7. Detekteres som typer eller differentielt splejsede isoformer af disse værktøjer (Ding et al. 2017).,
Visualisering
Der er mange bioinformatik værktøjer til visualisering af RNA-seq data, herunder genom-browsere, såsom ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Genom-Kort, Savant, værktøjer er specielt designet til RNA-seq data, såsom RNAseqViewer, samt nogle pakker til differentiel gen-ekspression analyse, der muliggør visualisering, som DESeq2 og DEXseq i Bioconductor. Pakker, såsom cummerbund og sashimi plots, er også udviklet til visualisering-eksklusive formål.,
Funktionelle Profilering
Det seneste skridt i en standard, transcriptomics undersøgelse er generelt karakterisering af molekylære funktioner eller veje, hvor differentielt udtrykte gener er involveret. Gen ontologi, Bioconductor, DAVID eller Babelomics indeholder annotationsdata for de fleste modelarter, som kan bruges til funktionel annotation. Som for nye udskrifter, protein-kodende udskrifter kan være funktionelt kommenteret hjælp orthology med hjælp af databaser såsom SwissProt, Pfam, og InterPro., Gene Ontology (GO) giver mulighed for en vis udskiftelighed af funktionel information på tværs af ortologer. Blast2GO er et populært værktøj, der tillader massiv annotation af komplet transkriptome mod en række databaser og kontrollerede ordforråd. Rfam-databasen indeholder de fleste velkarakteriserede RNA-familier, der kan bruges til funktionel annotation af lange ikke-kodende RNA ‘ er.
avanceret analyse
den avancerede analyse af RNA-se seq inkluderer normalt andre RNA-se.og integration med andre teknologier, som er skitseret i figur 4., Mere information om anvendelser af RNA-se., se venligst Denne artikel anvendelser af RNA-se..
figur 3. Den avancerede analyse af RNA-se.data.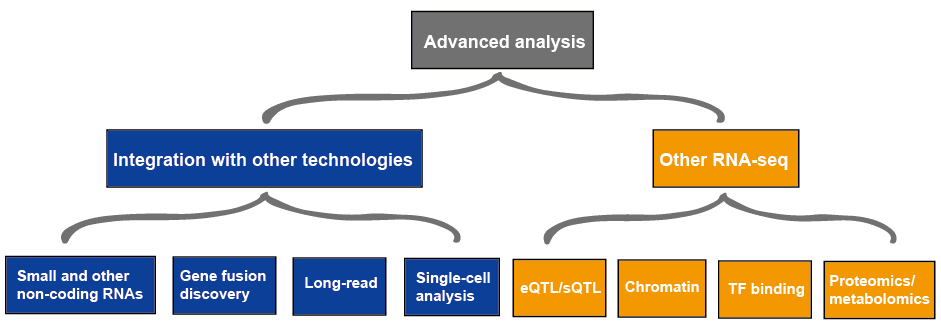
Vores erfarne bioinformatik forskere er dygtige i at udnytte de avancerede bioinformatiske værktøjer til at håndtere de mange sekvenser, der genereres af den næste og tredje generation sequencing. Vi leverer både sekventering og bioinformatik til genomforskning, transkriptomik, epigenomik, mikrobiel genomik, single-cell sekventering og PacBio SMRT sekventering.