RNA-sekvenování (RNA-seq) má širokou škálu aplikací a neexistuje optimální potrubí pro všechny případy. Přezkoumáváme všechny hlavní kroky v analýze dat RNA-seq, včetně kontroly kvality, zarovnání čtení, kvantifikace hladin genů a přepisů, diferenciální genová exprese, funkční profilování a pokročilá analýza. Budou diskutovány později.
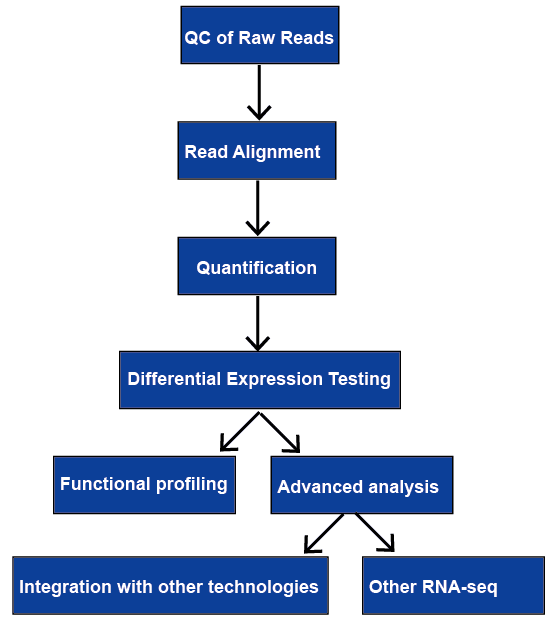
Obrázek 1. Obecný pracovní postup analýzy RNA-seq.,
kontrola kvality raw reads
kontrola kvality RNA-seq raw reads se skládá z analýzy kvality sekvence, obsahu GC, obsahu adaptéru, přepsaných k-mers a duplikovaných čtení, věnovaných detekci sekvenčních chyb, kontaminací a artefaktů PCR. Kvalita čtení klesá ke konci čtení 3, základny s nízkou kvalitou, proto by měly být odstraněny, aby se zlepšila mapovatelnost., Kromě kvality surových dat, kontrolu kvality syrového čte také zahrnuje analýzu číst zarovnání (číst jednotnost a GC obsahu), kvantifikace (3′ zaujatost, biotopů, a low-počítá) a reprodukovatelnosti (korelace, analýza hlavních komponent, a dávkové účinky).
Tabulka 1. Nástroje pro kontrolu kvality RNA-seq raw čte.,
| Tools | Applications |
| NGSQC | Quality control of raw reads generated by Illumina platforms. |
| FastQC | Quality control of raw reads generated by any platforms., |
| FASTX-Toolkit | Discard of low-quality reads, trim adaptor sequences, and elimination of poor quality bases. |
| Trimmonmatic | |
| Picard | Quality control in read alignment, including the determination of read uniformity and GC content., |
| RSeQC | |
| Qualimap | |
| NOISeq | Provide useful plots for quality control of count data. |
| EDASeq |
Read alignment
There are generally three strategies for read alignment, genome mapping, transcriptome mapping, and de novo assembly., Bez ohledu na to, zda je k dispozici odkaz na genom nebo transkriptom, může reads jedinečně mapovat nebo být přiřazen k více pozicím v odkazu, které jsou označovány jako multi-mapované čtení nebo vícenásobné čtení. Genomické multiready jsou obecně způsobeny opakovanými sekvencemi nebo sdílenými doménami paralogických genů. Transkriptomové multi-mapování vzniká častěji kvůli genovým isoformám. Identifikace a kvantifikace transkriptu jsou proto důležitými výzvami pro alternativně exprimované geny., Pokud odkaz není k dispozici, RNA-seq čte jsou sestaveny de novo pomocí balíčků, jako je SOAPdenovo-Trans, oázy, Trans-ABySS, nebo Trinity. Pe strand specifické a dlouhé délky čtení jsou preferovány, protože jsou více informativní. Vznikající technologie s dlouhým čtením, jako je sekvenování PacBio SMRT a sekvenování Nanopore, mohou generovat přepisy v plné délce pro většinu genů.
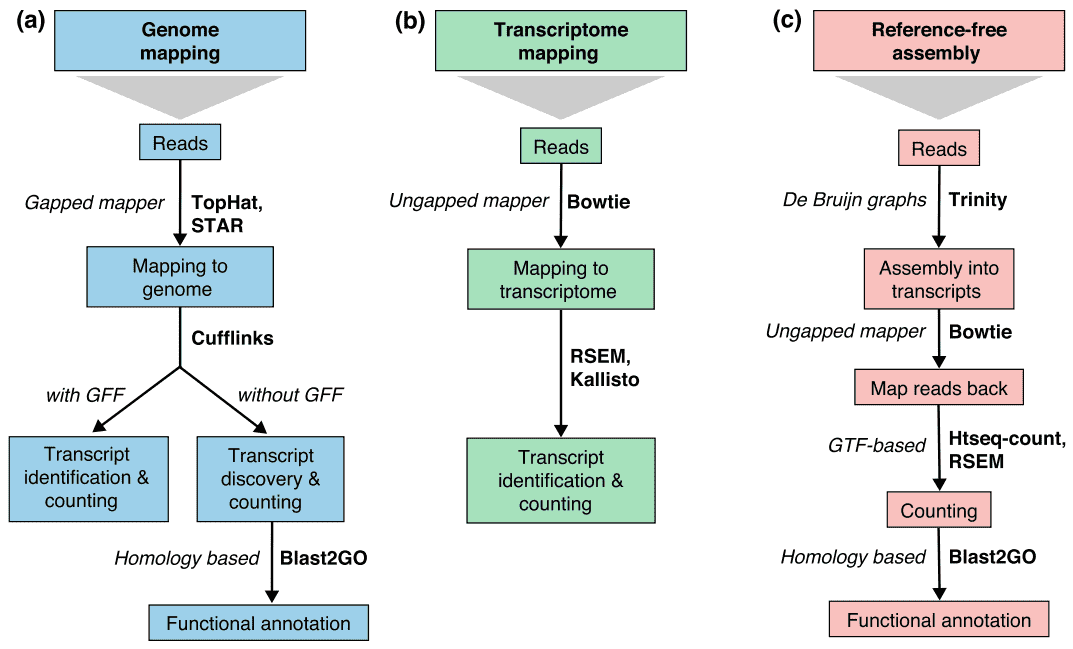
Obrázek 2. Tři základní strategie pro RNA-seq čtení mapování (Conesa et al . 2016)., Zkratky: GFF, obecný formát funkcí; GTF, formát přenosu genů; RSEM, RNA-seq podle maximalizace očekávání.
Tabulka 2. Srovnání genomových a de novo montážních strategií pro RNA-seq analýzu.,lignment of reads to known splice site is not required
- More computational intense
- Sensitive to sequencing error
Table 3., The public sources of RNA-seq data.
Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.
Table 4. The common tools for transcript quantification.
| Tools | Principles and Applications |
| TopHat | Using an expectation-maximization approach that estimates transcript abundances., |
| manžetové knoflíčky | určené k využití čtení PE a mohou používat informace GTF k identifikaci vyjádřených přepisů nebo mohou odvodit přepisy de novo pouze z mapovacích dat. |
| RSEM | kvantifikovat výraz z mapování transkriptomu. přidělit multi-mapování čte mezi přepisem a výstupem v rámci vzorku normalizovaných hodnot korigovaných pro sekvenční zkreslení., |
| eXpress | |
| Sailfish | |
| kallisto | |
| NURD | Provides an efficient way of estimating transcript expression from SE reads with a low memory and computing cost. |
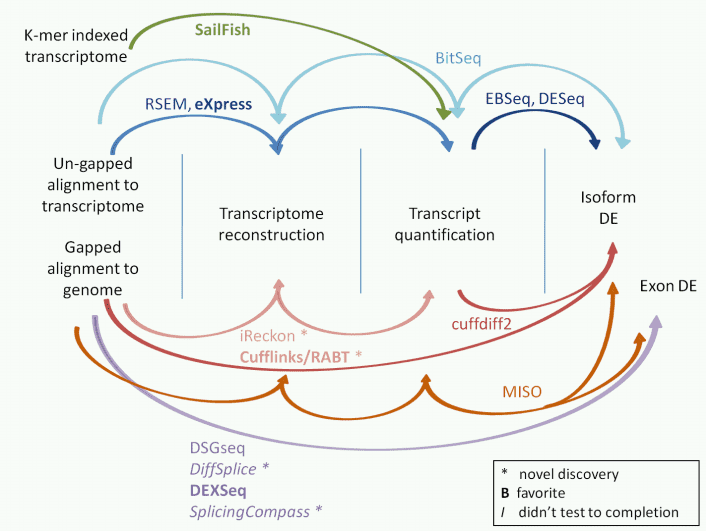
Figure 3. The tools for isoform expression quantification.,
testování diferenciální exprese
testování diferenciální exprese se používá k vyhodnocení, zda je jeden gen odlišně exprimován v jednom stavu ve srovnání s druhým(s). Normalizační metody je třeba přijmout před porovnáním různých vzorků. RPKM a TPM normalizují nejdůležitější faktor, sekvenční hloubku. TMM, DESeq a UpperQuartile mohou ignorovat vysoce variabilní a / nebo vysoce vyjádřené funkce., Další faktory, které interferují s uvnitř vzorku srovnání zahrnovat přepis délka, poziční předsudky v pokrytí, průměrná velikost fragmentu, a GC obsahu, což může být normalizována pomocí nástrojů, jako je DESeq, rozhrnovacích, baySeq, a NOISeq. Dávkové efekty mohou být stále přítomny po normalizaci, které mohou být minimalizovány vhodným experimentálním designem nebo odstraněny metodami, jako je boj nebo ARSyN.
Tabulka 5. Normalizační nástroje pro diferenciální expresní testování.,r>Quantile
TMM
RPKM
TMM
Upperquartile
Alternative splicing analysis
Alternative splicing (AS) is a posttranscriptional process which generates different transcripts from the same gene and is vital in response to environmental stimuli by producing diverse protein products., Bylo vyvinuto několik nástrojů bioinformatiky pro detekci jako z experimentálních dat. Srovnání těchto detekčních nástrojů pomocí dat RNA-seq bylo provedeno společností Ding v roce 2017 a výsledky jsou uvedeny v tabulce 7. Prokázali, že TopHat a jeho následný nástroj, FineSplice, jsou nejrychlejšími nástroji, zatímco těstoviny jsou nejpomalejší program. Kromě toho může AltEventFinder detekovat nejvyšší počet křižovatek a RSR detekuje nejnižší počet křižovatek. Jiné nástroje, jako je TopHat, pravděpodobně detekují falešně pozitivní., Ze dvou nástrojů, které detekují differentially spliced isoforms, je rMATS rychlejší než rSeqDiff, ale detekuje méně odlišně spojované isoformy než rSeqDiff.
Tabulka 7. Detekovány jako typy nebo různě sestříhané izoformy těchto nástrojů (Ding et al. 2017).,
Vizualizace
Existuje mnoho bioinformatika nástroje pro vizualizaci RNA-seq dat, včetně genomu prohlížeče, jako je ReadXplorer, UCSC browser, Integrative Genomics Viewer (IGV), Genomové Mapy, Učenec, nástroje speciálně určené pro RNA-seq dat, jako RNAseqViewer, stejně jako některé balíčky pro diferenciální genové exprese analýzy, které umožňují vizualizace, jako DESeq2 a DEXseq v Bioconductor. Balíčky, jako jsou pozemky CummeRbund a Sashimi, byly také vyvinuty pro exkluzivní účely vizualizace.,
funkční profilování
posledním krokem ve standardní transkriptomické studii je obecně charakterizace molekulárních funkcí nebo cest, do kterých jsou zapojeny diferenciálně exprimované geny. Genová ontologie, Biokonduktor, DAVID nebo Babelomika obsahují anotační údaje pro většinu modelových druhů, které lze použít pro funkční anotaci. Pokud jde o nové přepisy, přepisy kódování proteinů lze funkčně anotovat pomocí ortologie pomocí databází, jako jsou SwissProt, Pfam a InterPro., Genová ontologie (GO) umožňuje určitou vyměnitelnost funkčních informací napříč ortology. Blast2GO je populární nástroj, který umožňuje masivní anotaci kompletního transkriptomu proti různým databázím a kontrolovaným slovníkům. Databáze Rfam obsahuje většinu dobře charakterizovaných RNA rodin, které lze použít pro funkční anotaci dlouhých nekódujících RNA.
pokročilá analýza
pokročilá analýza RNA-seq obvykle zahrnuje další RNA-seq a integraci s jinými technologiemi, která je popsána na obrázku 4., Více informací o aplikacích RNA-seq naleznete v tomto článku aplikace RNA-Seq.
obrázek 3. Pokročilá analýza dat RNA-seq.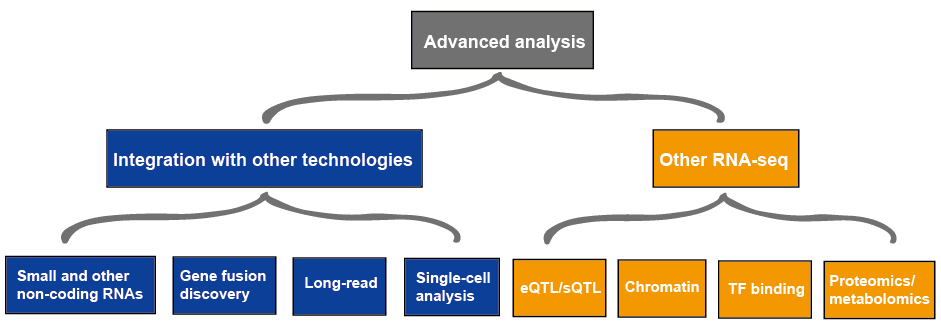
Náš zkušený bioinformatika vědci jsou kvalifikovaní v s využitím pokročilých bioinformatických nástrojů, jak se vypořádat s mnoha sekvence generované druhé a třetí generace sekvenování. Poskytujeme jak sekvenování a bioinformatiky služby pro genomiku, transcriptomics, epigenomics, mikrobiální genomika, single-cell sekvenování, a PacBio SMRT sekvenování.